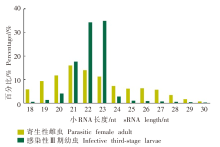
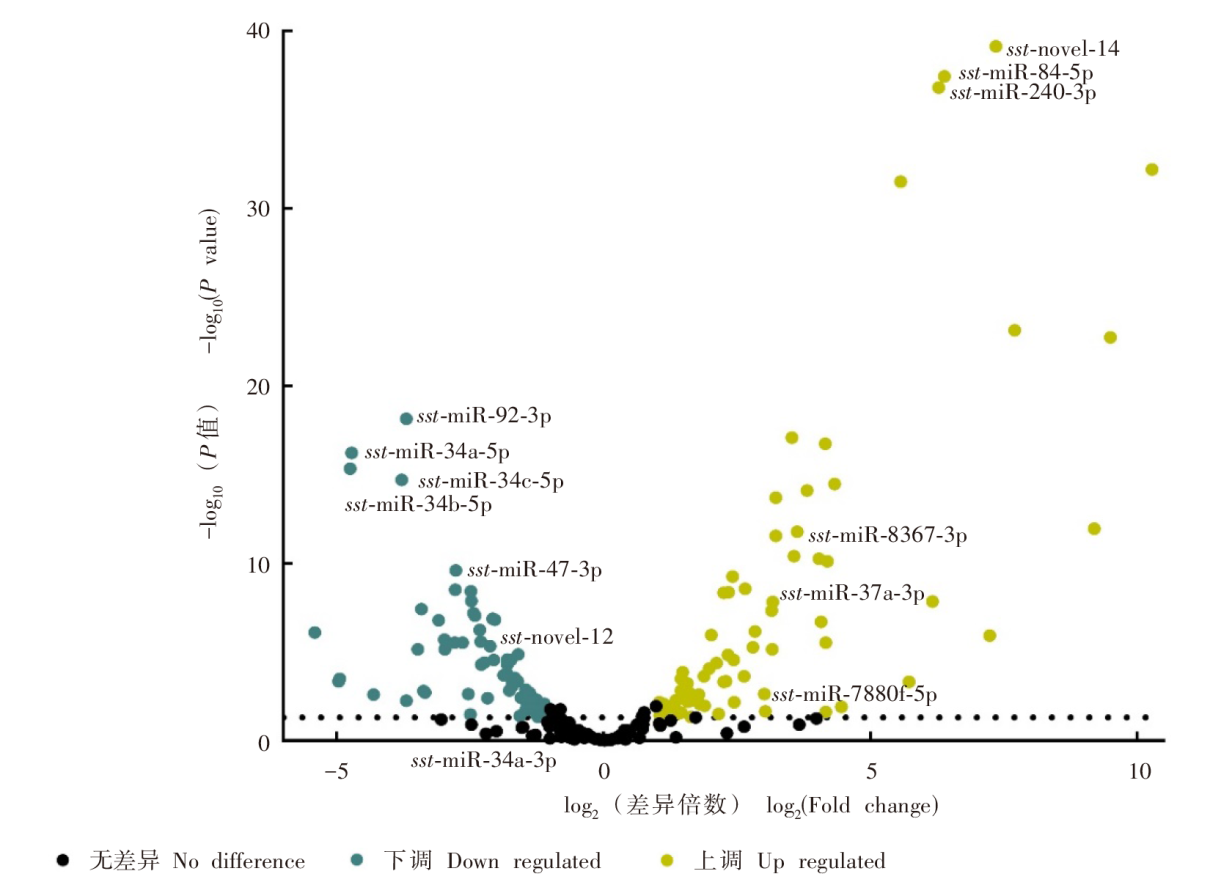
| [1] | Beknazarova M, Whiley H, Ross K. Strongyloidiasis: a disease of socioeconomic disadvantage[J]. Int J Environ Res Public Health, 2016, 13(5): 517. | | [2] | Olsen A, van Lieshout L, Marti H, et al. Strongyloidiasis: the most neglected of the neglected tropical diseases?[J]. Trans R Soc Trop Med Hyg, 2009, 103(10): 967-972. | | [3] | Puthiyakunnon S, Boddu S, Li YJ, et al. Strongyloidiasis: an insight into its global prevalence and management[J]. PLoS Negl Trop Dis, 2014, 8(8): e3018. | | [4] | Hu JY. The establishment of Strongyloides stercoralis infected geril model and initial attempt to establish CRISPR/Cas9 knockout method[D]. Wuhan: Huazhong Agricultural University, 2018: 2. (in Chinese) | | | (胡锦阳. 粪类圆线虫感染沙鼠模型的建立及CRISPR/Cas9基因敲除方法的初步尝试[D]. 武汉: 华中农业大学, 2018: 2.) | | [5] | Marcos LA, Terashima A, Dupont HL, et al. Strongyloides hyperinfection syndrome: an emerging global infectious disease[J]. Trans R Soc Trop Med Hyg, 2008, 102(4): 314-318. | | [6] | Starr MC, Montgomery SP. Soil-transmitted helminthiasis in the United States: a systematic review: 1940—2010[J]. Am J Trop Med Hyg, 2011, 85(4): 680-684. | | [7] | Vasquez-Rios G, Pineda-Reyes R, Pineda-Reyes J, et al. Strongyloides stercoralis hyperinfection syndrome: a deeper understanding of a neglected disease[J]. J Parasit Dis, 2019, 43(2): 167-175. | | [8] | Hammond SM. An overview of microRNAs[J]. Adv Drug Deliv Rev, 2015, 87: 3-14. | | [9] | Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene Lin-4 encodes small RNAs with antisense complementarity to Lin-14[J]. Cell, 1993, 75(5): 843-854. | | [10] | Pasquini G, Kunej T. A map of the microRNA regulatory networks identified by experimentally validated microRNA-target interactions in five domestic animals: cattle, pig, sheep, dog, and chicken[J]. OMICS, 2019, 23(9): 448-456. | | [11] | Song XW, Li Y, Cao XF, et al. microRNAs and their regulatory roles in plant-environment interactions[J]. Annu Rev Plant Biol, 2019, 70: 489-525 | | [12] | Ulusan Ba?c? ?, Caner A. The role of microRNAs in parasitology[J]. Turkiye Parazitol Derg, 2020, 44(2): 102-108. | | [13] | Ahmed R, Chang ZS, Younis AE, et al. Conserved miRNAs are candidate post-transcriptional regulators of developmental arrest in free-living and parasitic nematodes[J]. Genome Biol Evol, 2013, 5(7): 1246-1260. | | [14] | Ma GX, Luo YF, Zhu HH, et al. microRNAs of Toxocara canis and their predicted functional roles[J]. Parasit Vectors, 2016, 9: 229. | | [15] | Winter AD, Weir W, Hunt M, et al. Diversity in parasitic nematode genomes: the microRNAs of Brugia pahangi and Haemonchus contortus are largely novel[J]. BMC Genomics, 2012, 13: 4. | | [16] | Xu MJ, Fu JH, Nisbet AJ, et al. Comparative profiling of microRNAs in male and female adults of Ascaris suum[J]. Parasitol Res, 2013, 112(3): 1189-1195. | | [17] | Pomari E, Malerba G, Veschetti L, et al. Identification of miRNAs of Strongyloides stercoralis L1 and iL3 larvae isolated from human stool[J]. Sci Rep, 2022, 12(1): 9957. | | [18] | Zhang Y. Genome-wide identfication and characterization of novel LncRNAs and extracellular vesicle preliminary study in Strongyloides stercoralis[D]. Wuhan: Huazhong Agricultural University, 2019: 17. (in Chinese) | | | (张映. 粪类圆线虫lncRNA的鉴定和验证以及细胞外囊泡的初步研究[D]. 武汉: 华中农业大学, 2019: 17.) | | [19] | Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome[J]. Genome Biol, 2009, 10(3): R25. | | [20] | Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data[J]. Bioinformatics, 2010, 26(1): 139-140. | | [21] | Hunt VL, Tsai IJ, Coghlan A, et al. The genomic basis of parasitism in the Strongyloides clade of nematodes[J]. Nat Genet, 2016, 48(3): | | [22] | Britton C, Laing R, Devaney E. Small RNAs in parasitic nematodes-forms and functions[J]. Parasitology, 2020, 147(8): 855-864. | | [23] | Liu N, Landreh M, Cao KJ, et al. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila[J]. Nature, 2012, 482(7386): 519-523. | | [24] | Yang JR, Chen DP, He YN, et al. miR-34 modulates Caenorhabditis elegans lifespan via repressing the autophagy gene atg9[J]. Age, 2013, 35(1): 11-22. | | [25] | Isik M, Blackwell TK, Berezikov E. microRNA mir-34 provides robustness to environmental stress response via the DAF-16 network in C. elegans[J]. Sci Rep, 2016, 6: 36766. | | [26] | Boulias K, Horvitz HR. The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO[J]. Cell Metab, 2012, 15(4): 439-450. | | [27] | Zhang XC, Zabinsky R, Teng YD, et al. microRNAs play critical roles in the survival and recovery of Caenorhabditis elegans from starvation-induced L1 diapause[J]. Proc Natl Acad Sci USA, 2011, 108(44): 17997-18002. | | [28] | Pérez MG, Spiliotis M, Rego N, et al. Deciphering the role of miR-71 in Echinococcus multilocularis early development in vitro[J]. PLoS Negl Trop Dis, 2019, 13(12): e0007932. | | [29] | Zheng YD, Guo XL, He W, et al. Effects of Echinococcus multilocularis miR-71 mimics on murine macrophage RAW264.7 cells[J]. Int Immunopharmacol, 2016, 34: 259-262. | | [30] | Yang ML, Wang YL, Jiang F, et al. miR-71 and miR-263 jointly regulate target genes chitin synthase and chitinase to control locust molting[J]. PLoS Genet, 2016, 12(8): e1006257. | | [31] | Davis MW, Birnie AJ, Chan AC, et al. A conserved metalloprotease mediates ecdysis in Caenorhabditis elegans[J]. Development, 2004, 131(23): 6001-6008. | | [32] | Gamble HR, Purcell JP, Fetterer RH. Purification of a 44 kilodalton protease which mediates the ecdysis of infective Haemonchus contortus larvae[J]. Mol Biochem Parasitol, 1989, 33(1): 49-58. | | [33] | Stepek G, McCormack G, Birnie AJ, et al. The astacin metalloprotease moulting enzyme NAS-36 is required for normal cuticle ecdysis in free-living and parasitic nematodes[J]. Parasitology, 2011, 138(2): 237-248. | | [34] | Audhya A, Desai A, Oegema K. A role for Rab5 in structuring the endoplasmic reticulum[J]. J Cell Biol, 2007, 178(1): 43-56. | | [35] | Sann SB, Crane MM, Lu H, et al. Rabx-5 regulates RAB-5 early endosomal compartments and synaptic vesicles in C. elegans[J]. PLoS One, 2012, 7(6): e37930. | | [36] | Kamikura DM, Cooper JA. Clathrin interaction and subcellular localization of Ce-DAB-1, an adaptor for protein secretion in Caenorhabditis elegans[J]. Traffic, 2006, 7(3): 324-336. |
|

 ), 周彩显1, 鲁志刚1,2, 张碧瀛1, 周涛勋1, 胡敏1,*(
), 周彩显1, 鲁志刚1,2, 张碧瀛1, 周涛勋1, 胡敏1,*(