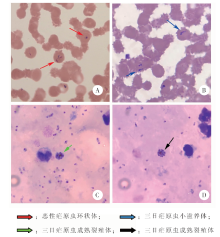
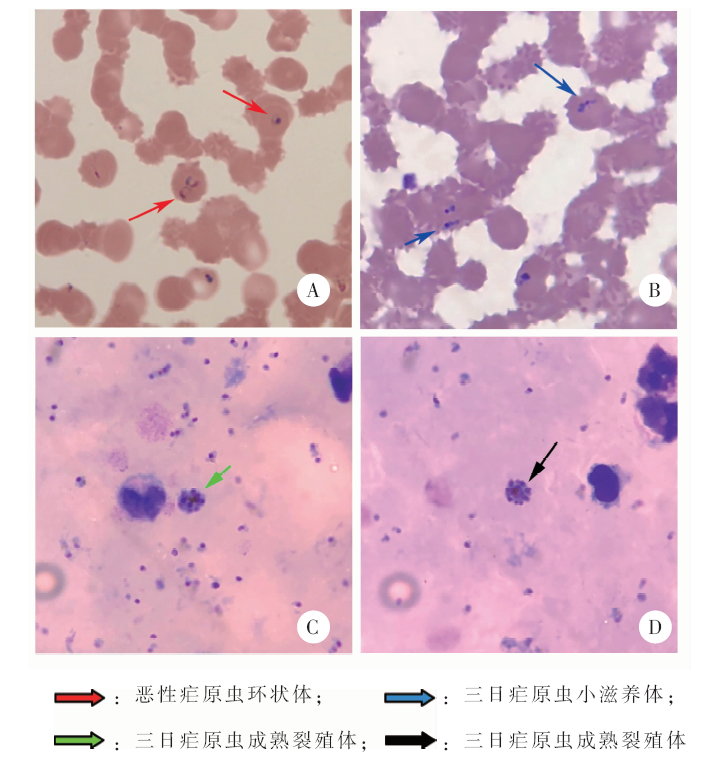
| [1] | Liu J, Sun Y, Shi W, et al., The first imported case of Rift Valley fever in China reveals a genetic reassortment of different viral lineages[J]. Emerg Microbes Infect, 2017, 6(1): e4. | | [2] | Cui S, Pan Y, Lyu Y, et al. Detection of yellow fever virus genomes from four imported cases in China[J]. Int J Infect Dis, 2017, 60: 93-95. | | [3] | 潘阳, 崔淑娟, 陈丽娟, 等. 我国首例输入性裂谷热病例病毒全基因组测序分析[J]. 国际病毒学杂志, 2017, 24(1): 1-4. | | [4] | 张丽, 丰俊, 张少森, 等. 2017年全国消除疟疾进展及疫情特征分析[J]. 中国寄生虫学与寄生虫病杂志, 2018, 36(3): 201-209. | | [5] | Wolkowicz T.The utility and perspectives of NGS-based methods in BSL-3 and BSL-4 laboratory-sequencing and analysis strategies[J]. Briefings Funct Genom, 2018, 17(6): 471-476. | | [6] | 李多, 康显虎, 赵晓楠, 等. 云南省2例人感染H5N6禽流感病毒的基因特征研究[J]. 国际病毒学杂志, 2018, 25(6): 361-364. | | [7] | José Antonio Garrido-Cardenas, Lilia González-Cerón, Manzano-Agugliaro F, et al. Plasmodium genomics: an approach for learning about and ending human malaria[J]. Parasitol Res, 2019, 118(1): 1-27. | | [8] | 何纬, 石莹, 田绿波, 等. 四川口岸首例输入性三日疟病例分析[J]. 中国病原生物学杂志, 2017, 12(8): 77-79. | | [9] | Huson DH, Beier SN, Flade I, et al. Megan community edition-interactive exploration and analysis of large-scale microbiome sequencing data[J]. PLoS Comput Biol, 2016, 12(6): e1004957. | | [10] | Huson DH, Albrecht B, Baĝci C, et al. MEGAN-LR: new algorithms allow accurate binning and easy interactive exploration of metagenomic long reads and contigs[J]. Biol Direct, 2018, 13(1): 6. | | [11] | 巫学兰. 疟原虫镜检及鉴定方法探讨[J]. 实验与检验医学, 2011, 29(2): 200-201. | | [12] | Moody A.Rapid diagnostic tests for malaria parasites[J]. Clin Microbiol Rev, 2002, 15(1): 66-78. | | [13] | Wang B, Han SS, Cho C, et al. Comparison of microscopy, nested-PCR, and real-time PCR assays using high-throughput screening of pooled samples for diagnosis of malaria in asymptomatic carriers from areas of endemicity in Myanmar[J]. J Clin Microbiol, 2014, 52(6): 1838-1845. |
|

 ), Xiao-mei WANG, Yu-lan SUN, Yan-ning LV, Xiang-feng DOU, Quan-yi WANG*(
), Xiao-mei WANG, Yu-lan SUN, Yan-ning LV, Xiang-feng DOU, Quan-yi WANG*(