中国寄生虫学与寄生虫病杂志 ›› 2021, Vol. 39 ›› Issue (1): 27-35.doi: 10.12140/j.issn.1000-7423.2021.01.004
乌兰图雅1( ), 殷旭红1, 崔云虹1, 刘丹1, 王亭富1, 苗雨润1, 阿木日汗1, 曹民治2, 赵志伟3, 邢方超3, 鲁建英3, 高娃1,*()
), 殷旭红1, 崔云虹1, 刘丹1, 王亭富1, 苗雨润1, 阿木日汗1, 曹民治2, 赵志伟3, 邢方超3, 鲁建英3, 高娃1,*()
Wulantuya1(), YIN Xu-hong1, CUI Yun-hong1, LIU Dan1, WANG Ting-fu1, MIAO Yu-run1, 1, CAO Min-zhi2, ZHAO Zhi-wei3, XING Fang-chao3, LU Jian-ying3, GAO Wa1,*()
摘要:
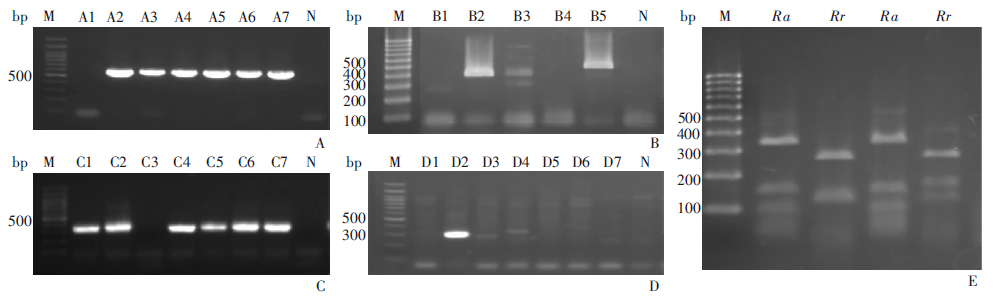
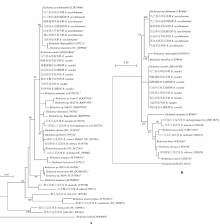
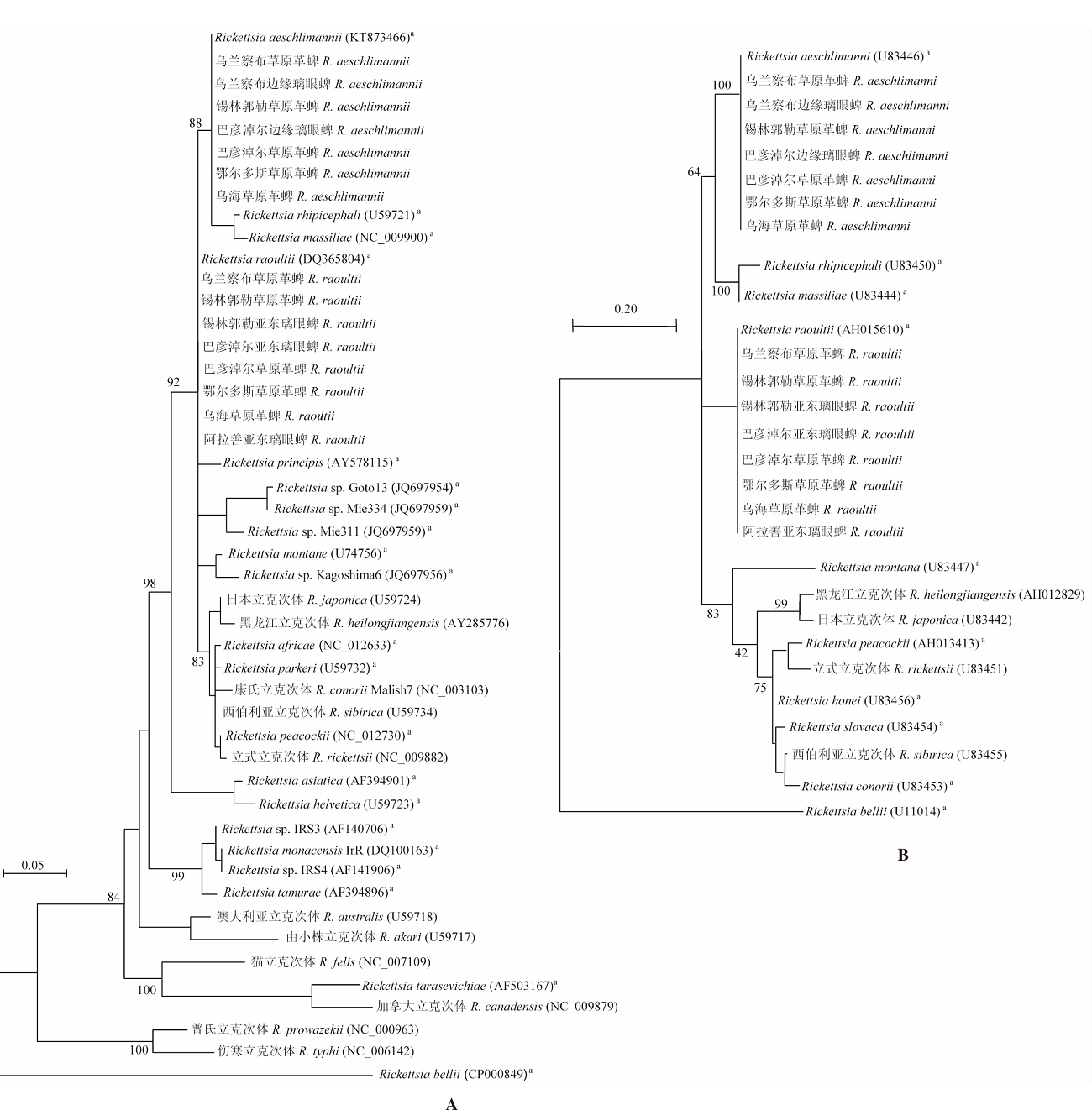
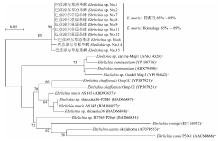
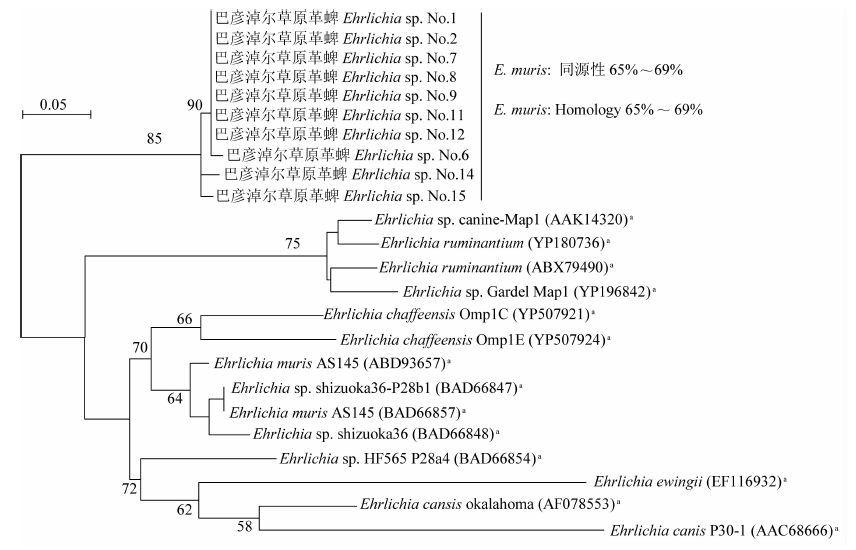
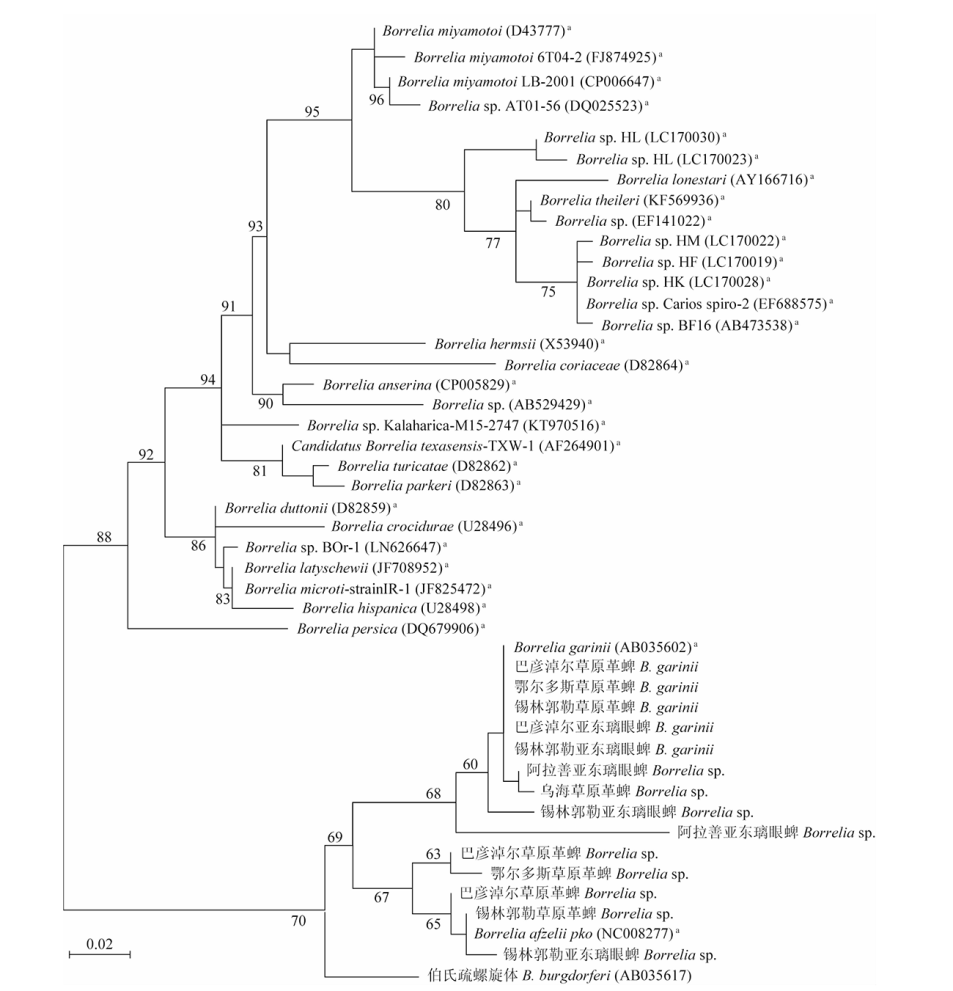
目的 了解内蒙古中西部草原蜱类的群落结构、携带病原体多样性及基因型。 方法 于2016—2019年春夏季,在内蒙古中西部草原,采用动物体表搜集法采集蜱标本,进行蜱种鉴定。解剖摘取蜱的唾液腺并提取基因组DNA,以斑点热立克次体柠檬酸合成酶A(gltA)、疏螺旋体以鞭毛蛋白B(flaB)、埃立克体属以外膜蛋白质1(omp1)、无形体属主要表面蛋白2(msp2)为靶基因进行PCR扩增初筛。立克次体gltA初筛阳性样品经限制性片段长度多态性(RFLP)分类,再根据蜱种和地区每类选20~30个代表性样品进行gltA、立克次体外膜蛋白A(rOmpA)基因测序。扩增序列测序后用BLAST、Clustal W和MEGA 7.0软件进行同源性分析,以邻接法构建系统进化树。 结果 共采集成蜱3 822只,经形态学特征和特异性18S rRNA基因分型法鉴定,隶属于2属3种,分别为草原革蜱、亚东璃眼蜱和边缘璃眼蜱,其中草原革蜱占55.7%(2 129/3 822)、亚东璃眼蜱占30.0%(1 147/3 822),为该地区的优势蜱种。PCR检测结果显示,立克次体gltA基因阳性蜱1 899只,阳性率为49.7%(1 899/3 822),gltA基因阳性样品根据RFLP结果分为两类,两类样品的gltA基因序列均为581 bp,与R. raoultii(DQ365804)或R. aeschlimanni(KT873466)的同源性为100%;两类样品的rOmpA基因均长367 bp,与R. raoultii(AH015610)或R. aeschlimanni(U83466)的同源性为100%,与gltA基因的结果相符。3 822只蜱中,R. raoultii和R. aeschlimanni的阳性率分别为37.2%(1 422/3 822)和12.5%(477/3 822),其中草原革蜱中分别为58.5%(1 245/2 129)和11.1%(477/2 129);亚东璃眼蜱中分别为15.4%(177/1 147)和0;边缘璃眼蜱中分别为0和44.0%(240/546)。疏螺旋体flaB基因阳性蜱28只,阳性率为0.7%(28/3 822),其中草原革蜱中为0.8%(16/2 129),亚东璃眼蜱为1.0%(12/1 147)。共获得疏螺旋体flaB基因序列10条,与莱姆病主要病原体B. garinii(AB035602)和B. afzelii PKo(NC008277)的同源性分别为90.6%~100%和95.6%~100%。亚东璃眼蜱中B. garinii和B. afzelii的阳性率分别为0.9%(10/1 147)和0.2%(2/1 147),草原革蜱中均为0.4%(8/2 129)。3 822只蜱中omp1基因阳性1只,TA克隆后获得8个氨基酸序列相同的克隆和3个氨基酸序列存在差异的克隆,11个克隆的氨基酸序列与E. muris的同源性最高,但仅为65%~69%。3 822只蜱中均未检出无形体属菌群。系统进化树分析结果显示,3种蜱感染的立克次体均与R. raoultii和R. aeschlimanni聚在一簇。在获得的10条疏螺旋体菌群flaB基因序列中,源于草原革蜱和亚东璃眼蜱的1条序列与B. garinii聚在一簇,草原革蜱的另1条序列与B. afzelii聚在一簇,其余8条序列与B. garinii和B. afzelii的flaB基因序列处在不同的分支。草原革蜱感染的埃立克体属菌与目前已知的埃立克体属菌群关系较远,形成独自的聚类。 结论 内蒙古中西部草原存在草原革蜱、亚东璃眼蜱和边缘璃眼蜱,蜱类中广泛存在斑点热立克次体和莱姆病螺旋体的感染,是R. raoultii、R. aeschlimanni、B. garinii和B. afzelii潜在的自然疫源地。有必要加强该地区蜱媒传染病的预防控制工作。
中图分类号: