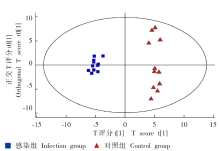
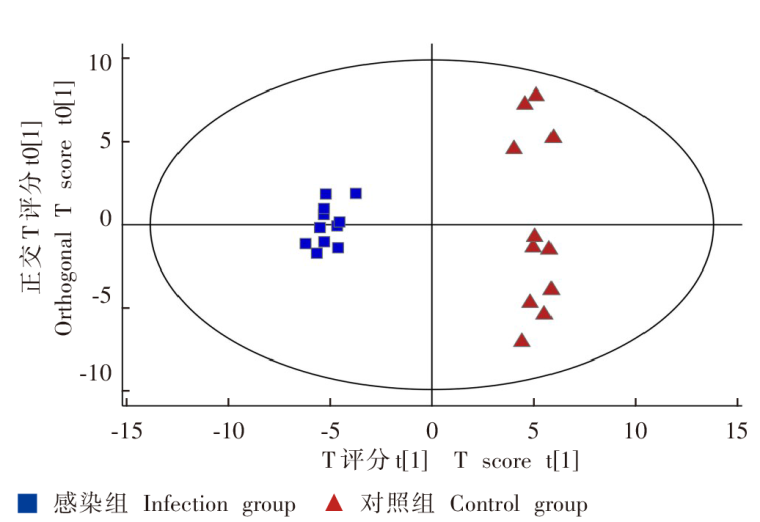
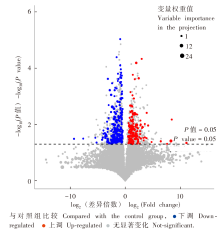
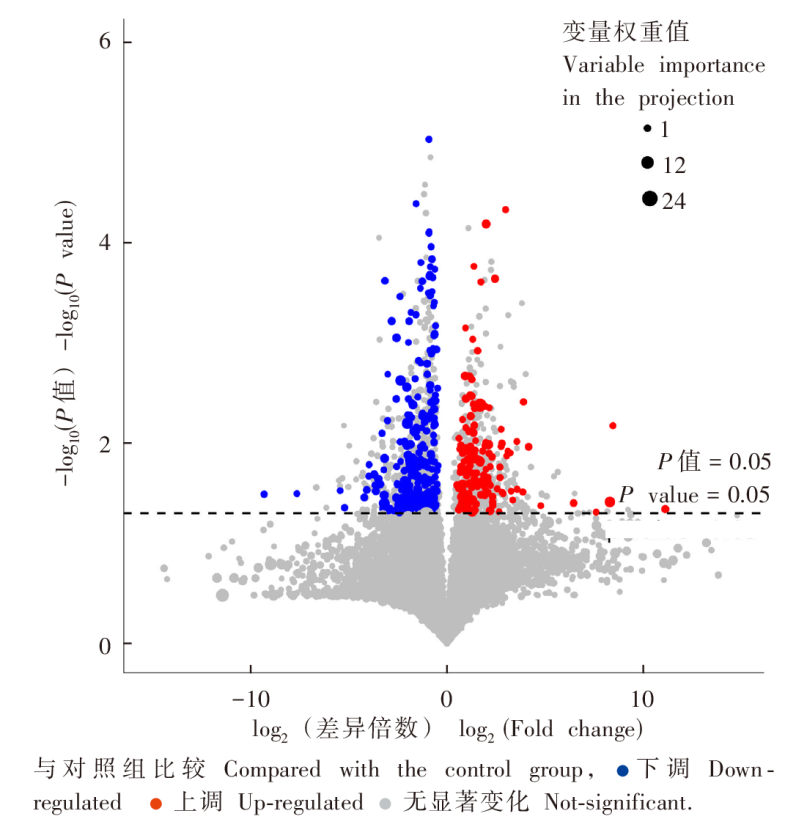
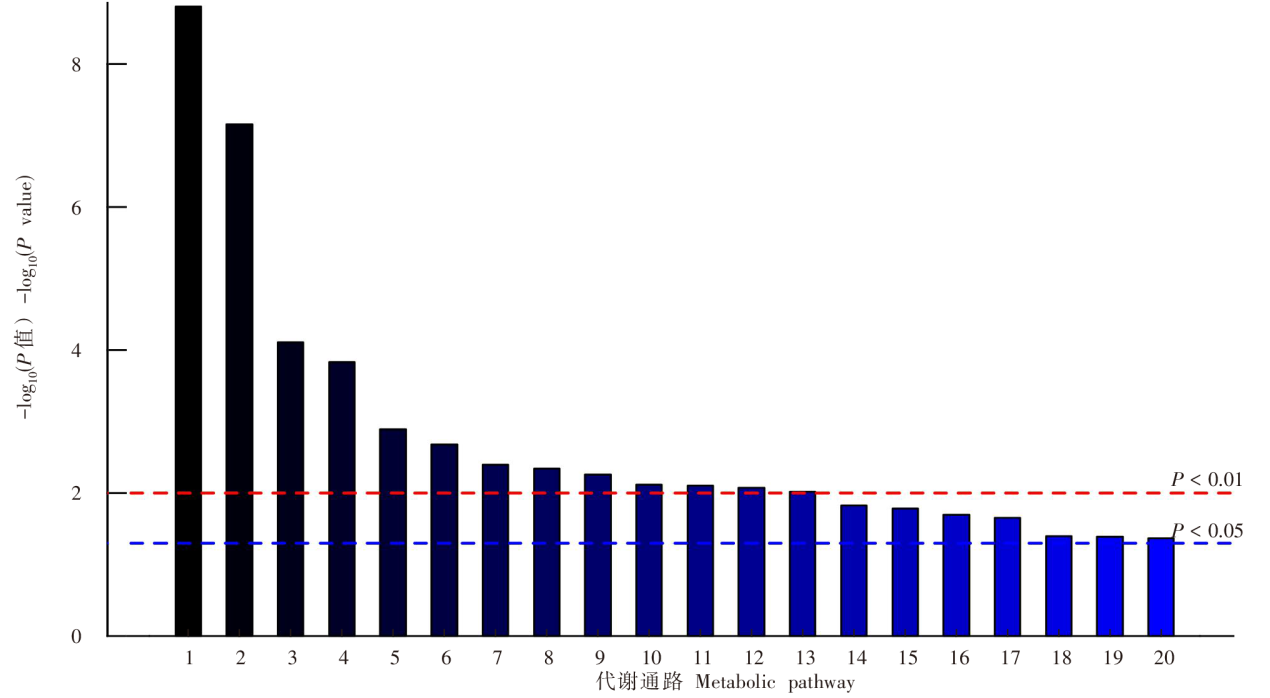
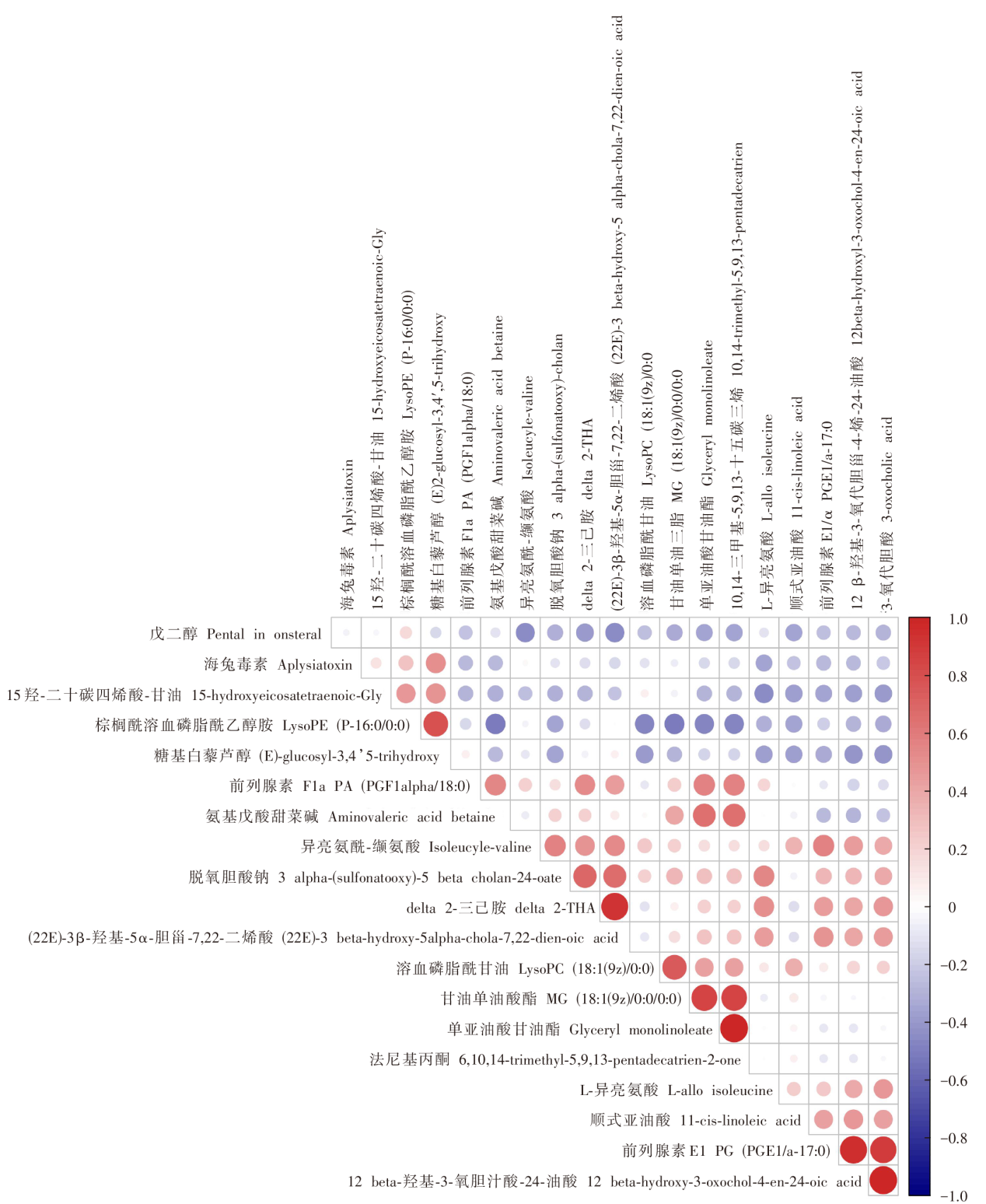
| [1] |
Zhang Y, Zhang SQ. Gut microbiome and health[M]. Beijing: China Light Industry Press, 2021: 1. (in Chinese)
|
|
(张彦, 张双庆. 肠道微生物组与健康[M]. 北京: 中国轻工业出版社, 2021: 1.)
|
| [2] |
Ni J, Wu GD, Albenberg L, et al. Gut microbiota and IBD: causation or correlation?[J]. Nat Rev Gastroenterol Hepatol, 2017, 14(10): 573-584.
doi: 10.1038/nrgastro.2017.88
pmid: 28743984
|
| [3] |
Singer-Englar T, Barlow G, Mathur R. Obesity, diabetes, and the gut microbiome: an updated review[J]. Expert Rev Gastroenterol Hepatol, 2019, 13(1): 3-15.
doi: 10.1080/17474124.2019.1543023
|
| [4] |
Christovich A, Luo XM. Gut microbiota, leaky gut, and autoimmune diseases[J]. Front Immunol, 2022, 13: 946248.
doi: 10.3389/fimmu.2022.946248
|
| [5] |
Witkowski M, Weeks TL, Hazen SL. Gut microbiota and cardiovascular disease[J]. Circ Res, 2020, 127(4): 553-570.
doi: 10.1161/CIRCRESAHA.120.316242
pmid: 32762536
|
| [6] |
Park EM, Chelvanambi M, Bhutiani N, et al. Targeting the gut and tumor microbiota in cancer[J]. Nat Med, 2022, 28(4): 690-703.
doi: 10.1038/s41591-022-01779-2
pmid: 35440726
|
| [7] |
Sampson TR, Debelius JW, Thron T, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease[J]. Cell, 2016, 167(6): 1469-1480.e12.
doi: 10.1016/j.cell.2016.11.018
|
| [8] |
Nikolova VL, Smith MRB, Hall LJ, et al. Perturbations in gut microbiota composition in psychiatric disorders: a review and meta-analysis[J]. JAMA Psychiatry, 2021, 78(12): 1343-1354.
doi: 10.1001/jamapsychiatry.2021.2573
pmid: 34524405
|
| [9] |
Ling ZX, Liu X, Cheng YW, et al. Gut microbiota and aging[J]. Crit Rev Food Sci Nutr, 2022, 62(13): 3509-3534.
doi: 10.1080/10408398.2020.1867054
|
| [10] |
Parker A, Romano S, Ansorge R, et al. Fecal microbiota transfer between young and aged mice reverses hallmarks of the aging gut, eye, and brain[J]. Microbiome, 2022, 10(1): 68.
doi: 10.1186/s40168-022-01243-w
pmid: 35501923
|
| [11] |
Coman V, Vodnar DC. Gut microbiota and old age: modulating factors and interventions for healthy longevity[J]. Exp Gerontol, 2020, 141: 111095.
doi: 10.1016/j.exger.2020.111095
|
| [12] |
Zeng T, Lü S, Tian LG, et al. Temporal trends in disease burden of major human parasitic diseases in China from 1990 to 2019[J]. Chin J Schisto Control, 2023, 35(1): 7-14, 37. (in Chinese)
|
|
(曾婷, 吕山, 田利光, 等. 1990—2019年我国主要人体寄生虫病疾病负担变化趋势研究[J]. 中国血吸虫病防治杂志, 2023, 35(1): 7-14.)
|
| [13] |
Motran CC, Silvane L, Chiapello LS, et al. Helminth infections: recognition and modulation of the immune response by innate immune cells[J]. Front Immunol, 2018, 9: 664.
doi: 10.3389/fimmu.2018.00664
pmid: 29670630
|
| [14] |
Llinás-Caballero K, Caraballo L. Helminths and bacterial microbiota: the interactions of two of humans’ “old friends”[J]. Int J Mol Sci, 2022, 23(21): 13358.
doi: 10.3390/ijms232113358
|
| [15] |
Lund ME, Greer J, Dixit A, et al. A parasite-derived 68-mer peptide ameliorates autoimmune disease in murine models of type 1 diabetes and multiple sclerosis[J]. Sci Rep, 2016, 6: 37789.
doi: 10.1038/srep37789
pmid: 27883079
|
| [16] |
Cantacessi C, Giacomin P, Croese J, et al. Impact of experimental hookworm infection on the human gut microbiota[J]. J Infect Dis, 2014, 210(9): 1431-1434.
doi: 10.1093/infdis/jiu256
pmid: 24795483
|
| [17] |
Mangiola F, Nicoletti A, Gasbarrini A, et al. Gut microbiota and aging[J]. Eur Rev Med Pharmacol Sci, 2018, 22(21): 7404-7413.
|
| [18] |
O’Toole PW, Jeffery IB. Gut microbiota and aging[J]. Science, 2015, 350(6265): 1214-1215.
doi: 10.1126/science.aac8469
pmid: 26785481
|
| [19] |
Kim S, Jazwinski SM. The gut microbiota and healthy aging: amini-review[J]. Gerontology, 2018, 64(6): 513-520.
doi: 10.1159/000490615
|
| [20] |
Bravo JA, Forsythe P, Chew MV, et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve[J]. Proc Natl Acad Sci USA, 2011, 108(38): 16050-16055.
doi: 10.1073/pnas.1102999108
|
| [21] |
Kim CS, Cha LN, Sim M, et al. Probiotic supplementation improves cognitive function and mood with changes in gut microbiota in community-dwelling older adults: a randomized, double-blind, placebo-controlled, multicenter trial[J]. J Gerontol A Biol Sci Med Sci, 2021, 76(1): 32-40.
doi: 10.1093/gerona/glaa090
|
| [22] |
Webster HC, Gamino V, Andrusaite AT, et al. Tissue-based IL-10 signalling in helminth infection limits IFNγ expression and promotes the intestinal Th2 response[J]. Mucosal Immunol, 2022, 15(6): 1257-1269.
doi: 10.1038/s41385-022-00513-y
pmid: 35428872
|
| [23] |
Hu ZH, Zhang CL, Sifuentes-Dominguez L, et al. Small proline-rich protein 2A is a gut bactericidal protein deployed during helminth infection[J]. Science, 2021, 374(6568): eabe6723.
|
| [24] |
Su CW, Chen CY, Jiao LF, et al. Helminth-induced and Th2-dependent alterations of the gut microbiota attenuate obesity caused by high-fat diet[J]. Cell Mol Gastroenterol Hepatol, 2020, 10(4): 763-778.
doi: S2352-345X(20)30105-3
pmid: 32629118
|
| [25] |
Shimokawa C, Kato T, Takeuchi T, et al. CD8+ regulatory T cells are critical in prevention of autoimmune-mediated diabetes[J]. Nat Commun, 2020, 11(1): 1922.
doi: 10.1038/s41467-020-15857-x
pmid: 32321922
|
| [26] |
Franco-Lopez J, Duplessis M, Bui A, et al. Correlations between the composition of the bovine microbiota and vitamin B12 abundance[J]. mSystems, 2020, 5(2): e00107-e00120.
|
| [27] |
Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients[J]. Proc Natl Acad Sci USA, 2008, 105(43): 16731-16736.
doi: 10.1073/pnas.0804812105
|
| [28] |
Bush JR, Alfa MJ. Increasing levels of Parasutterella in the gut microbiome correlate with improving low-density lipoprotein levels in healthy adults consuming resistant potato starch during a randomised trial[J]. BMC Nutr, 2020, 6(1): 72.
doi: 10.1186/s40795-020-00398-9
|
| [29] |
Chen YJ, Wu H, Wu SD, et al. Parasutterella, in association with irritable bowel syndrome and intestinal chronic inflammation[J]. J Gastroenterol Hepatol, 2018, 33(11): 1844-1852.
doi: 10.1111/jgh.2018.33.issue-11
|
| [30] |
Rodríguez-Medina N, Barrios-Camacho H, Duran-Bedolla J, et al. Klebsiella variicola: an emerging pathogen in humans[J]. Emerg Microbes Infect, 2019, 8(1): 973-988.
doi: 10.1080/22221751.2019.1634981
|
| [31] |
Zafar H, Saier MH Jr. Gut Bacteroides species in health and disease[J]. Gut Microbes, 2021, 13(1): 1-20.
|
| [32] |
Iglesias-Vázquez L, van Ginkel Riba G, Arija V, et al. Composition of gut microbiota in children with autism spectrum disorder: a systematic review and meta-analysis[J]. Nutrients, 2020, 12(3): 792.
doi: 10.3390/nu12030792
|
| [33] |
Sakamoto M, Ikeyama N, Toyoda A, et al. Dialister hominis sp. nov., isolated from human faeces[J]. Int J Syst Evol Microbiol, 2020, 70(1): 589-595.
doi: 10.1099/ijsem.0.003797
|
| [34] |
Kong QM, Wang BT, Tian PJ, et al. Daily intake of Lactobacillus alleviates autistic-like behaviors by ameliorating the 5-hydroxytryptamine metabolic disorder in VPA-treated rats during weaning and sexual maturation[J]. Food Funct, 2021, 12(6): 2591-2604.
doi: 10.1039/D0FO02375B
|
| [35] |
Chang TT, Chen JW. Direct CCL4 inhibition modulates gut microbiota, reduces circulating trimethylamine N-oxide, and improves glucose and lipid metabolism in high-fat-diet-induced diabetes mellitus[J]. J Inflamm Res, 2021, 14: 6237-6250.
doi: 10.2147/JIR.S343491
|
| [36] |
Liang JQ, Li T, Nakatsu G, et al. A novel faecal Lachnoclostridium marker for the non-invasive diagnosis of colorectal adenoma and cancer[J]. Gut, 2020, 69(7): 1248-1257.
doi: 10.1136/gutjnl-2019-318532
|
| [37] |
Loukas A, Hotez PJ, Diemert D, et al. Hookworm infection[J]. Nat Rev Dis Primers, 2016, 2: 16088.
doi: 10.1038/nrdp.2016.88
pmid: 27929101
|
| [38] |
Gazzinelli-Guimaraes PH, Nutman TB. Helminth parasites and immune regulation[J]. F1000Res, 2018, 7: F1000FacultyRev-F1000Faculty1685.
|
| [39] |
Loukas A, Maizels RM, Hotez PJ. The Yin and Yang of human soil-transmitted helminth infections[J]. Int J Parasitol, 2021, 51(13/14): 1243-1253.
doi: 10.1016/j.ijpara.2021.11.001
|

 ), 沈续航2, 邓国强1, 胡赛敏1, 汪飞3, 张玲玲4, 沈继龙5,*(
), 沈续航2, 邓国强1, 胡赛敏1, 汪飞3, 张玲玲4, 沈继龙5,*(