
CHINESE JOURNAL OF PARASITOLOGY AND PARASITIC DISEASES ›› 2024, Vol. 42 ›› Issue (4): 439-446.doi: 10.12140/j.issn.1000-7423.2024.04.003
• ORIGINAL ARTICLES • Previous Articles Next Articles
ZHANG Yu1( ), ZHANG Ke2, LIU Jiawei1, WANG Anqi1, TUAN Yong3, ZHANG Dong1, YAN Liping1, LI Kai1,*()
), ZHANG Ke2, LIU Jiawei1, WANG Anqi1, TUAN Yong3, ZHANG Dong1, YAN Liping1, LI Kai1,*()
Received:2023-12-15
Revised:2024-03-16
Online:2024-08-30
Published:2024-08-22
Contact:
E-mail: Supported by:CLC Number:
ZHANG Yu, ZHANG Ke, LIU Jiawei, WANG Anqi, TUAN Yong, ZHANG Dong, YAN Liping, LI Kai. Metagenomic analysis and potential assessment of Hyalomma asiaticum in the distribution area of Przewalski’s horses[J]. CHINESE JOURNAL OF PARASITOLOGY AND PARASITIC DISEASES, 2024, 42(4): 439-446.
Add to citation manager EndNote|Ris|BibTeX
URL: https://www.jsczz.cn/EN/10.12140/j.issn.1000-7423.2024.04.003

Fig. 1
The morphological and molecular identification of H. aciaticum A: Male H. aciaticum; B: Female H. aciaticum; C, D: PCR electrophoresis for COⅠ sequence. M: DNA marker; 1-24: DNA of male tick; 25-48: DNA of female tick.
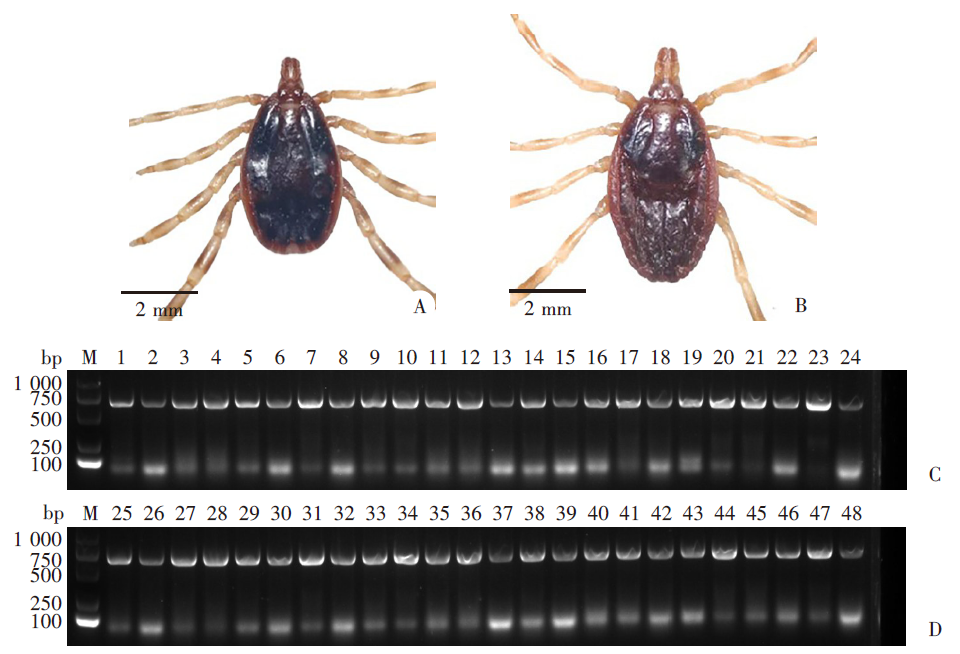
Table 1
Diversity index of the microbial community structure of male and female H. asiaticum
| 性别 Gender | 细菌群落丰度 Bacterial community abundance | 病毒群落丰度 Viral community abundance | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 门水平 Phylum level | 种水平 Species level | 门水平 Phylum level | 种水平 Species level | |||||||
| 香农指数 Shannon index | 辛普森指数 Simpson index | 香农指数 Shannon index | 辛普森指数 Simpson index | 香农指数 Shannon index | 辛普森指数 Simpson index | 香农指数 Shannon index | 辛普森指数 Simpson index | |||
| 雄性 Male | 0.836 | 0.474 | 3.551 | 0.073 | 0.965 | 0.456 | 0.901 | 0.479 | ||
| 雌性 Female | 0.859 | 0.482 | 4.058 | 0.108 | 0.982 | 0.470 | 0.982 | 0.486 | ||
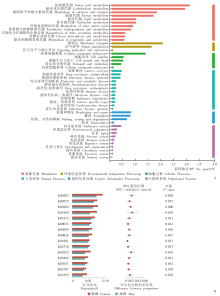
Fig. 2
KEGG pathway annotation of H. asiaticum (A) and differential expression pathway between male and female (B)
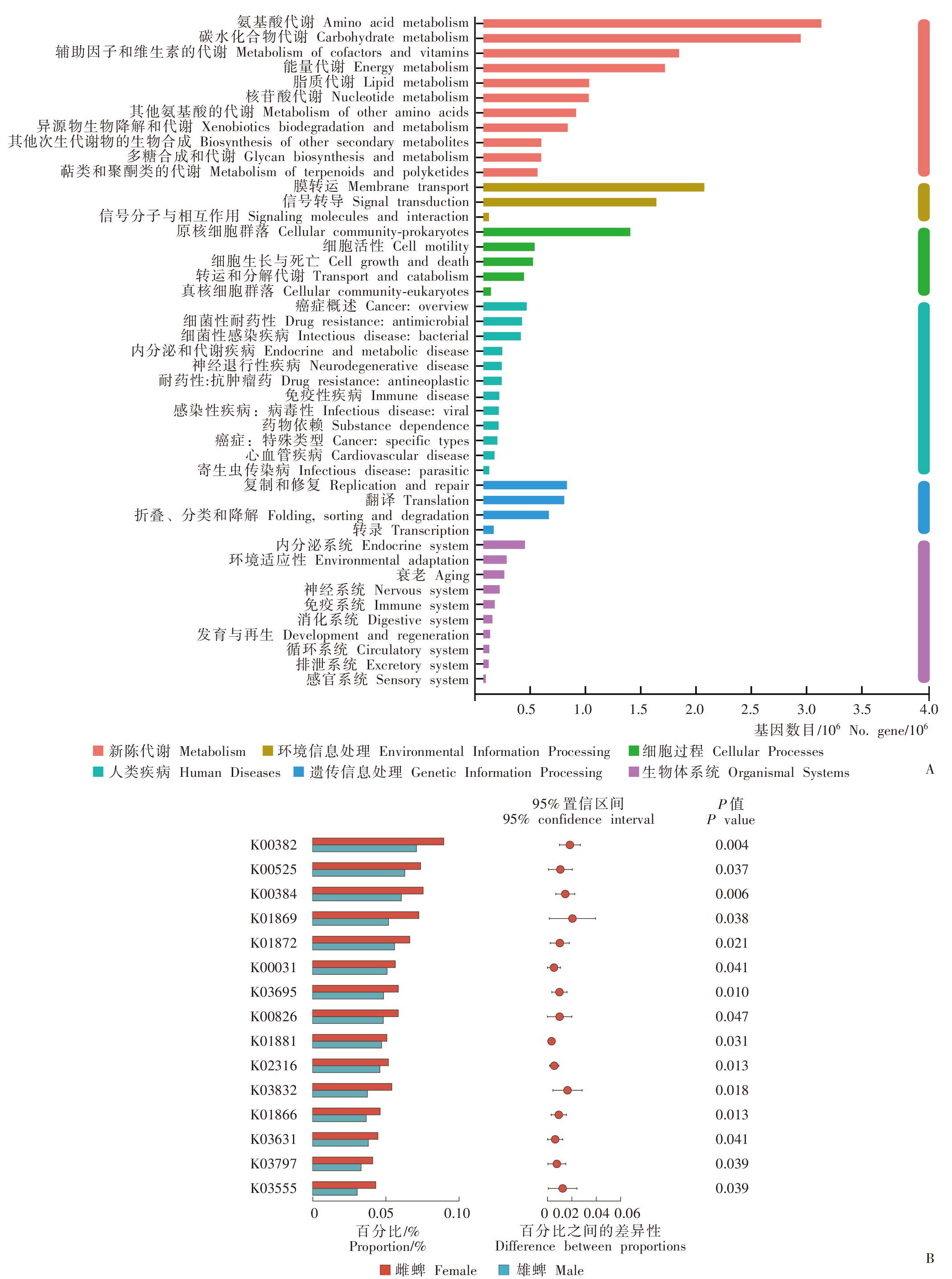
Table 2
Antibiotic resistance functions annotation results of H. asiaticum
| 抗性基因类型 Resistance gene types | 丰度/% Abundance/% | 抗生素类型 Antibiotic type | 类别说明 Category description |
|---|---|---|---|
| Baca | 62.53 | 杆菌肽 Bacitracin | 抑制细菌细胞壁合成,具有抗菌、抗炎、抗氧化、降血脂、降血糖等多种作用 Inhibits bacterial cell wall synthesis and has antibacterial, anti-inflammatory, antioxidant, lipid-lowering and glucose-lowering effects |
| Mexf | 7.04 | 多重耐药 Multiple drug resistance | 抗性结瘤细胞分裂转运系统;耐多药外排泵 Resistant nodulation cell division transport system; multidrug efflux pump |
| Mexb | 3.69 | 多重耐药 Multiple drug resistance | 抗性结瘤细胞分裂转运系统;耐多药外排泵 Resistant nodulation cell division transport system; multidrug efflux pump |
| Ceob | 2.85 | 多重耐药 Multiple drug resistance | 抗性结瘤细胞分裂转运系统;耐多药外排泵 Resistant nodulation cell division transport system; multidrug efflux pump |
| Sul1 | 2.58 | 磺酰胺 Sulfonamide | 磺酰胺抗性的二氢酸合成酶;不能被磺酰胺抑制 Sulfonamide-resistant dihydropteroate synthase; cannot be inhibited by sulfonamide |
| Acrb | 2.24 | 多重耐药 Multiple drug resistance | 抗性结瘤细胞分裂转运系统;耐多药外排泵 Resistant nodulation cell division transport system; multidrug efflux pump |
| Bl1 fox | 1.79 | β-内酰胺 β-lactam | A类β-内酰胺酶;使分子的抗菌特性失活 Class A β-lactamase; deactivate the antibacterial properties of the molecule |
| Mexi | 1.67 | 多重耐药 Multiple drug resistance | 抗性结瘤细胞分裂转运系统;耐多药外排泵 Resistant nodulation cell division transport system; multidrug efflux pump |
| Ant2ia | 1.66 | 氨基糖苷类 Aminoglycosides | 氨基糖苷O-核苷酸转移酶;通过腺苷酸化修饰氨基糖苷 Aminoglycoside O-nucleotide transferase; modifies of aminoglycosides by adenylation |
| Bl3 cpha | 1.38 | β-内酰胺 | B类β-内酰胺酶;使分子的抗菌特性失活Class B β-lactamase; deactivates the antibacterial properties of the molecule |
| Emrd | 0.88 | 多重耐药 Multiple drug resistance | 耐多药外排泵 Multidrug efflux pump |
| Mexw | 0.83 | 多重耐药 Multiple drug resistance | 抗性结瘤细胞分裂转运系统;耐多药外排泵 Resistant nodulation cell division transport system; multidrug efflux pump |
| Bcr | 0.74 | - | - |
| Macb | 0.70 | 大环内酯 Macrolide | 抗性结瘤细胞分裂转运体系统;耐多药外排泵;大环内酯特异性外排系统 Resistant nodulation cell division transport system; multidrug efflux pump; macrolide specific efflux system |
| Mexd | 0.62 | 多重耐药 Multiple drug resistance | 抗性结瘤细胞分裂转运系统;耐多药外排泵 Resistant nodulation cell division transport system; multidrug efflux pump |
| [1] | Földvári G, Szabó É, Tóth GE, et al. Emergence of Hyalomma marginatum and Hyalommarufipes adults revealed by citizen science tick monitoring in Hungary[J]. Transbound Emerg Dis, 2022, 69(5): e2240-e2248. |
| [2] | Guglielmone AA, Robbins RG, Apanaskevich DA, et al. The hard ticks of the world[M]. Berlin: Springer, 2013: 649-681. |
| [3] | Valcárcel F, González J, González MG, et al. Comparative ecology of Hyalommalusitanicum and Hyalomma marginatum Koch, 1844 (Acarina ∶ Ixodidae)[J]. Insects, 2020, 11(5): 303. |
| [4] | Hu EC, Hu ZX, Mi XY, et al. Distribution prediction of Hyalomma asiaticum (Acari ∶ Ixodidae) in a localized region in Northwestern China[J]. J Parasitol, 2022, 108(4): 330-336. |
| [5] | Yu PF, Liu ZJ, Niu QL, et al. Molecular evidence of tick-borne pathogens in Hyalomma anatolicum ticks infesting cattle in Xinjiang Uygur Autonomous Region, Northwestern China[J]. Exp Appl Acarol, 2017, 73(2): 269-281. |
| [6] | Chitimia-Dobler L, Schaper S, Rieß R, et al. Imported Hyalomma ticks in Germany in 2018[J]. Parasit Vectors, 2019, 12(1): 134. |
| [7] | Benyedem H, Lekired A, Mhadhbi M, et al. First insights into the microbiome of Tunisian Hyalomma ticks gained through next-generation sequencing with a special focus on H. scupense[J]. PLoS One, 2022, 17(5): e0268172. |
| [8] | Zakham F, Albalawi AE, Alanazi AD, et al. Viral RNA metagenomics of Hyalomma ticks collected from dromedary camels in Makkah Province, Saudi Arabia[J]. Viruses, 2021, 13(7): 1396. |
| [9] | Qviller L, Viljugrein H, Loe LE, et al. The influence of red deer space use on the distribution of Ixodes ricinus ticks in the landscape[J]. Parasit Vectors, 2016, 9(1): 545. |
| [10] | McCoy KD, Léger E, Dietrich M. Host specialization in ticks and transmission of tick-borne diseases: a review[J]. Front Cell Infect Microbiol, 2013, 3: 57. |
| [11] |
Barker SC, Walker AR. Ticks of Australia. The species that infest domestic animals and humans[J]. Zootaxa, 2014(3816): 1-144.
doi: 10.11646/zootaxa.3816.1.1 pmid: 24943801 |
| [12] | Zhang Y, Liu JW, Zhang K, et al. Biological response to Przewalski’s horse reintroduction in native desert grasslands: a case study on the spatial analysis of ticks[J]. BMC Ecol Evol, 2024, 24(1): 61. |
| [13] | Almberg ES, Cross PC, Dobson AP, et al. Parasite invasion following host reintroduction: acase study of Yellowstone’s wolves[J]. Philos Trans R Soc Lond B Biol Sci, 2012, 367(1604): 2840-2851. |
| [14] | Zhang Y, Zhang K, Liu JW, et al. Spatial distribution and causation of Hyalomma ticks in the core area of Kalamaili Nature Reserve[J]. Acta Ecol Sin, 2024, 44(16): 1-13. (in Chinese) |
| (张钰, 张科, 刘佳伟, 等. 卡山保护区核心区璃眼蜱空间格局及成因分析[J]. 生态学报, 2024, 44(16): 1-13.) | |
| [15] | Bouquet J, Melgar M, Swei A, et al. Metagenomic-based surveillance of pacific coast tick Dermacentor occidentalis identifies two novel bunyaviruses and an emerging human ricksettsial pathogen[J]. Sci Rep, 2017, 7(1): 12234. |
| [16] |
Garrido-Cardenas JA, Manzano-Agugliaro F. The metagenomics worldwide research[J]. Curr Genet, 2017, 63(5): 819-829.
doi: 10.1007/s00294-017-0693-8 pmid: 28401295 |
| [17] |
Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection[J]. Annu Rev Pathol, 2019, 14: 319-338.
doi: 10.1146/annurev-pathmechdis-012418-012751 pmid: 30355154 |
| [18] | Ravi A, Ereqat S, Al-Jawabreh A, et al. Metagenomic profiling of ticks: identification of novel rickettsial genomes and detection of tick-borne canine parvovirus[J]. PLoS Negl Trop Dis, 2019, 13(1): e0006805. |
| [19] |
Gilbert L. The impacts of climate change on ticks and tick-borne disease risk[J]. Annu Rev Entomol, 2021, 66: 373-388.
doi: 10.1146/annurev-ento-052720-094533 pmid: 33417823 |
| [20] | Deng GF. Economic entomology of China. Vol.39, Acari: Ixodidae[M]. Beijing: Science Press, 1991: 295-317. (in Chinese) |
| (邓国藩. 中国经济昆虫志. 第39册, 蜱螨亚纲•硬蜱科[M]. 北京: 科学出版社, 1991: 295-317.) | |
| [21] | Yu X, Ye RY, Gong ZD. The ticks fauna of Xinjiang[M]. Xinjiang Science, Technology and Public Health Press, 1997: 8-74. (in Chinese) |
| (于心, 叶瑞玉, 龚正达. 新疆蜱类志[M]. 乌鲁木齐: 新疆科技卫生出版社, 1997: 8-74.) | |
| [22] | Yang XH, Zhao YQ, Chuai X, et al. The life cycle of Hyalomma scupense (Acari ∶ Ixodidae) under laboratory conditions[J]. Ticks Tick Borne Dis, 2022, 13(6): 102019. |
| [23] | Kumar B, Manjunathachar HV, Ghosh S. A review on Hyalomma species infestations on human and animals and progress on management strategies[J]. Heliyon, 2020, 6(12): e05675. |
| [24] | Batool M, Blazier JC, Rogovska YV, et al. Metagenomic analysis of individually analyzed ticks from Eastern Europe demonstrates regional and sex-dependent differences in the microbiota of Ixodes ricinus[J]. Ticks Tick Borne Dis, 2021, 12(5): 101768. |
| [25] | Rojas-Jaimes J, Lindo-Seminario D, Correa-Núñez G, et al. Characterization of the bacterial microbiome of Rhipicephalus (Boophilus) microplus collected from Pecari tajacu “Sajino” Madre de Dios, Peru[J]. Sci Rep, 2021, 11(1): 6661. |
| [26] | Xiang YL, Zhou JZ, Zhang Y, et al. Metagenomic analysis of Rhipicephalus microplus from minority autonomous prefectures in Guizhou Province, China[J]. Chin J Vector Biol Control, 2023, 34(3): 319-325. (in Chinese) |
|
(向昱龙, 周敬祝, 张燕, 等. 贵州省少数民族自治州微小扇头蜱的宏基因组分析[J]. 中国媒介生物学及控制杂志, 2023, 34(3): 319-325.)
doi: 10.11853/j.issn.1003.8280.2023.03.007 |
|
| [27] | Liu MC, Zhang JT, Chen JJ, et al. A global dataset of microbial community in ticks from metagenome study[J]. Sci Data, 2022, 9(1): 560. |
| [28] | Namina A, Kazarina A, Lazovska M, et al. Comparative microbiome analysis of three epidemiologically important tick species in Latvia[J]. Microorganisms, 2023, 11(8): 1970. |
| [29] | Gou HT, Xue HW, Yin H, et al. Phylogenetic study of Hyalomma spp. from China based on ITS and COⅠ genes[J]. Chin Vet Sci, 2016, 46(5): 563-567. (in Chinese) |
| (苟惠天, 薛慧文, 殷宏, 等. 基于ITS和COⅠ基因对于我国璃眼蜱的分类研究[J]. 中国兽医科学, 2016, 46(5): 563-567.) | |
| [30] | Yen YC, Kong LX, Lee L, et al. Characteristics of Crimean-Congo hemorrhagic fever virus (Xinjiang strain) in China[J]. Am J Trop Med Hyg, 1985, 34(6): 1179-1182. |
| [31] | Gui YJ, Shi S, Luo YJ, et al. Tick-borne diseases and tick control in Xinjiang[J]. Chin J Anim Infect Dis, 2023, 31(3): 213-220. (in Chinese) |
| (贵有军, 史深, 罗勇军, 等. 新疆蜱传疾病及蜱媒防制[J]. 中国动物传染病学报, 2023, 31(3): 213-220.) | |
| [32] |
Milholland MT, Xu G, Rich SM, et al. Pathogen coinfections harbored by adult Ixodes scapularis from white-tailed deer compared with questing adults across sites in Maryland, USA[J]. Vector Borne Zoonotic Dis, 2021, 21(2): 86-91.
doi: 10.1089/vbz.2020.2644 pmid: 33316206 |
| [33] | Civitello DJ, Rynkiewicz E, Clay K. Meta-analysis of co-infections in ticks[J]. Israel J EcolEvol, 2010, 56(3/4): 417-431. |
| [34] | Obregón D, Bard E, Abrial D, et al. Sex-specific linkages between taxonomic and functional profiles of tick gut microbiomes[J]. Front Cell Infect Microbiol, 2019, 9: 298. |
| [35] | Xu XL, Han AX, Ye SQ, et al. Metagenomic analysis of microbial community structure, antibiotic resistance genes, and virulence factors of ticks captured at Wenzhou Port, Zhejiang Province, China[J]. Chin J Vector Biol Control, 2021, 32(6): 763-771. (in Chinese) |
|
(许雪莲, 韩阿祥, 叶诗晴, 等. 温州口岸截获蜱体内微生物群落结构、抗生素抗性基因及毒力因子的宏基因组分析[J]. 中国媒介生物学及控制杂志, 2021, 32(6): 763-771.)
doi: 10.11853/j.issn.1003.8280.2021.06.021 |
|
| [36] | Zang S, Cao Q, Keremu A, et al. Food patch particularity and forging strategy of reintroduced Przewalski’s horse in North Xinjiang, China[J]. Turk J Zool, 2017, 41: 924-930. |
| [37] | Turghan MA, Jiang ZG, Niu ZZ. An update on status and conservation of the Przewalski’s horse (Equus ferus przewalskii): captive breeding and reintroduction projects[J]. Animals (Basel), 2022, 12(22): 3158. |
| [38] | Wang Y, Chu HJ, Han LL, et al. Factors affecting the home range of reintroduced Equus przewalskii in the Mt. Kalamaili Ungulate Nature Reserve[J]. Acta Ecol Sin, 2016, 36(2): 545-553. (in Chinese) |
| (王渊, 初红军, 韩丽丽, 等. 野放普氏野马(Equus przewalskii)家域面积及其影响因素[J]. 生态学报, 2016, 36(2): 545-553.) | |
| [39] | Zhang YJ, Zhang F, Cao Q, et al. Status and quality of water sources in the Kalamari Ungulate Nature Reserve: a case study in the released area of Equus przewalskii[J]. Arid Zone Res, 2014, 31(4): 665-671. (in Chinese) |
| (张永军, 张峰, 曹青, 等. 卡拉麦里山有蹄类自然保护区水源现状及水质分析: 以普氏野马放归区为例[J]. 干旱区研究, 2014, 31(4): 665-671.) | |
| [40] | Huang HQ, Zhang K, Zhang BR, et al. Analysis on the relationship between winter precipitation and the annual variation of horse stomach fly community in arid desert steppe, Northwest China (2007-2019)[J]. IntegrZool, 2022, 17(1): 128-138. |
| [41] | Zhang K, Zhang Y, Wang C, et al. Distribution characteristics of epidemic focus of Gasterophilus pecorum (Diptera ∶ Gasterophilidae) in the core habitat of released Przewalski’s horses[J]. Acta Ecol Sin, 2023, 43(14): 5840-5849. (in Chinese) |
| (张科, 张钰, 王臣, 等. 放归普氏野马核心区黑腹胃蝇疫源分布特点[J]. 生态学报, 2023, 43(14): 5840-5849.) | |
| [42] | Harrus S, Baneth G. Drivers for the emergence and re-emergence of vector-borne protozoal and bacterial diseases[J]. Int J Parasitol, 2005, 35(11/12): 1309-1318. |
| [43] | Young TP. Restoration ecology and conservation biology[J]. Biol Conserv, 2000, 92(1): 73-83. |
| [44] | Xia CJ, Cao J, Zhang HF, et al. Reintroduction of Przewalski’s horse (Equus ferus przewalskii) in Xinjiang, China: the status and experience[J]. Biol Conserv, 2014, 177: 142-147. |
| [1] | FEI Siwei, ZHAO Hanqing, YIN Jingxian, SUN Zhishan, GUO Xiaokui, KASSEGNE Kokouvi, ZHOU Xiaonong. Advances in the research development on ticks and tick-borne diseases: a bibliometric analysis [J]. CHINESE JOURNAL OF PARASITOLOGY AND PARASITIC DISEASES, 2023, 41(5): 609-618. |
| [2] | DUAN Yueli, YU Yiqi, ZHU Haoxiang, WANG Xinyu. A case of atypical cerebral cysticercosis [J]. CHINESE JOURNAL OF PARASITOLOGY AND PARASITIC DISEASES, 2023, 41(1): 128-130. |
| [3] | FU Sha-sha, HAN Chang-yu, WANG Xin-xiao, ZENG Ci-mei, WANG Xiao-qiang, OU Zong-xing. A case of pulmonary mixed infection of Strongyloides stercoralis and Legionella pneumophila [J]. CHINESE JOURNAL OF PARASITOLOGY AND PARASITIC DISEASES, 2022, 40(5): 686-688. |
| [4] | SONG Rui-qi, ZHAI Xue-jie, LI Cai-shan, GE Ting, GAN Lu, ZHANG Meng-yuan, FAN Xin-li, LI Yong-chang, ZHANG Yang, BAYIN Cha-han. Comparative analysis of biological characteristics of Hyalomma asiaticum and H. anatolicum Xinjiang isolates at different developmental stages [J]. CHINESE JOURNAL OF PARASITOLOGY AND PARASITIC DISEASES, 2022, 40(3): 369-378. |
| [5] | YAO Qing-mei, ZHOU Su-fang, ZHANG Yi, XIA Shang, XUE Jing-bo. Research progress on the correlations of tick-borne diseases with meteorological factors and their prevention measures in China [J]. CHINESE JOURNAL OF PARASITOLOGY AND PARASITIC DISEASES, 2020, 38(1): 123-127. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||