中国寄生虫学与寄生虫病杂志 ›› 2019, Vol. 37 ›› Issue (6): 727-729.doi: 10.12140/j.issn.1000-7423.2019.06.021
李仁清( ), 王小梅, 孙玉兰, 吕燕宁, 窦相峰, 王全意*()
), 王小梅, 孙玉兰, 吕燕宁, 窦相峰, 王全意*()
Ren-qing LI(), Xiao-mei WANG, Yu-lan SUN, Yan-ning LV, Xiang-feng DOU, Quan-yi WANG*()
摘要:
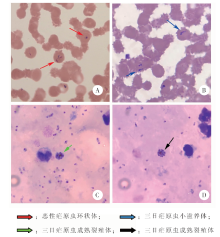
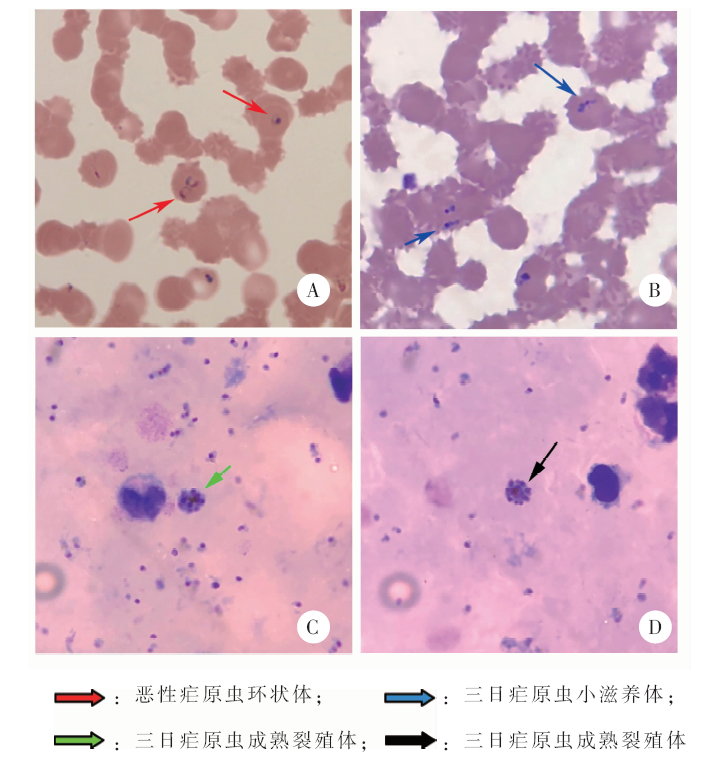
通过对1例自非洲输入的疟疾病例血液样本进行宏基因组二代测序(mNGS)检测,探讨mNGS在实验室检测疟疾上的可行性。采集患者外周血,制备厚、薄血膜,显微镜观察疟原虫感染情况。提取患者全血样本核酸,进行疟原虫核酸实时荧光PCR检测。将提取的患者全血样本核酸同步构建文库,采用Illumina Miseq二代测序仪进行mNGS检测。采用Megan6软件分析测序数据及重叠群序列的BLASTn比对结果,分配模式为对齐碱基模式。镜检结果显示,患者为恶性疟原虫和三日疟原虫混合感染,其中恶性疟原虫密度 ≥ 100 000/μl,三日疟原虫密度 ≥ 300/μl。实时荧光PCR检测结果显示,恶性疟原虫核酸阳性,间日疟、三日疟和卵形疟原虫核酸均为阴性。mNGS获得了271 526条测序数据,其中合格序列有254 674条。剔除与人类参考基因组匹配的序列后,最终得到1 833条测序数据,其中非人源数据只占总合格数据的0.7%。将上述序列拼接后,获得408条100个核苷酸以上长度的重叠群数据。Megan6分析结果显示,恶性疟原虫的测序数据分别占到总对齐碱基的44.7%(29 667/66 374),其重叠群数据占重叠群总对齐碱基的34.5%(22 885/66 376),因此恶性疟原虫是最优势的病原微生物。基于测序数据和重叠群数据,该病原可溯源到恶性疟原虫标准株3D7。
中图分类号: