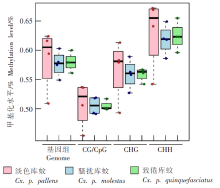
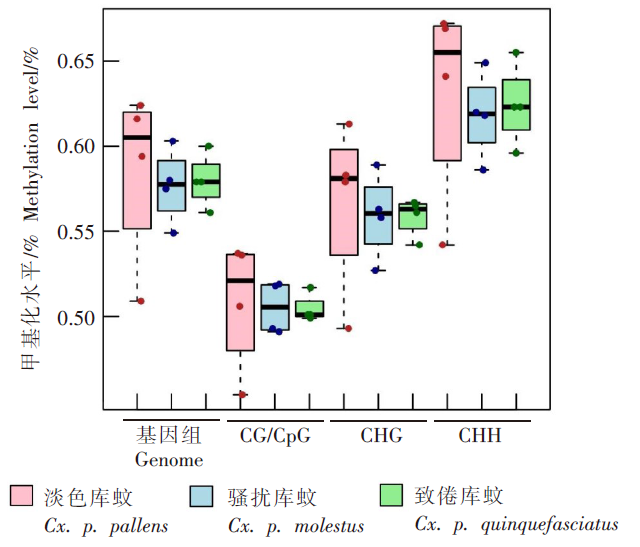
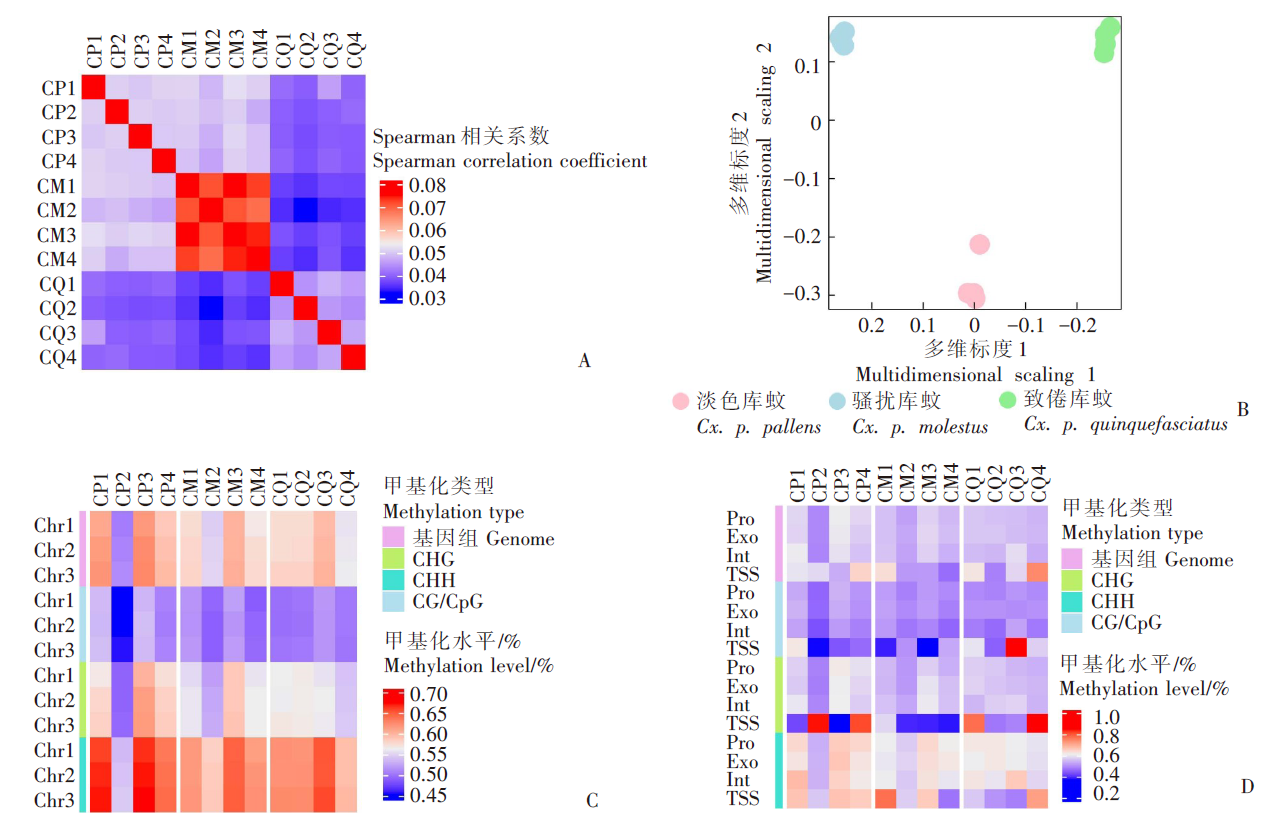
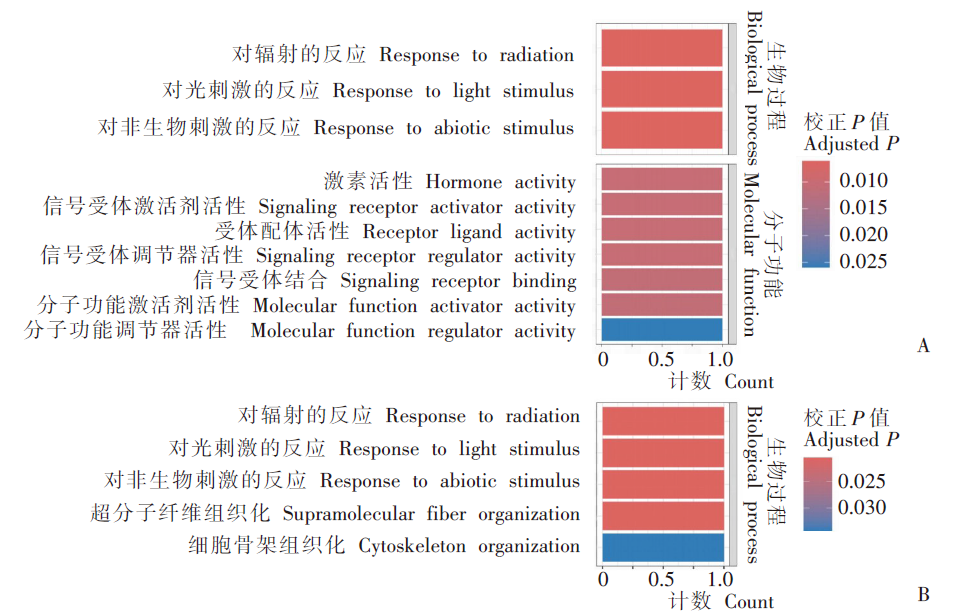
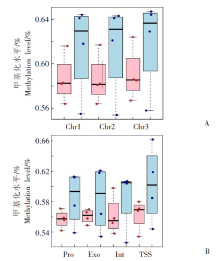
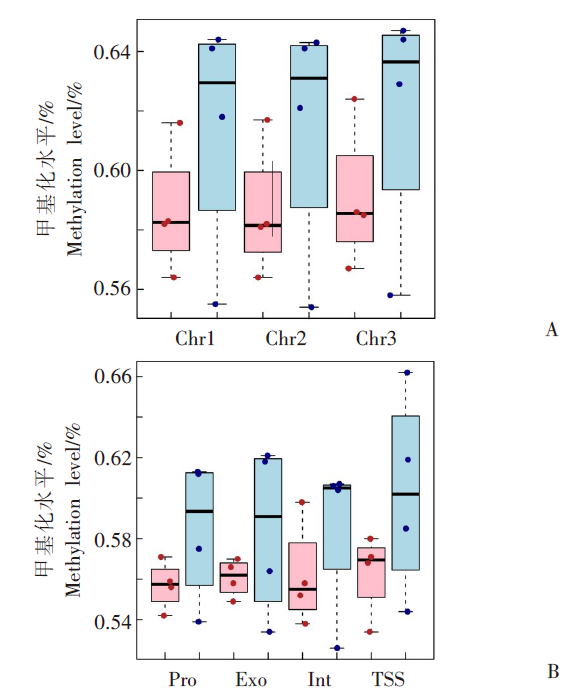
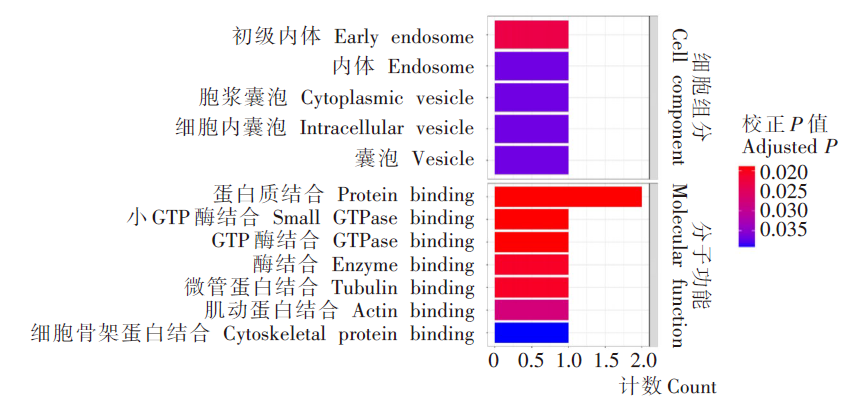
| [1] | Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information[J]. Nat Rev Genet, 2010, 11(4): 285-296. | | [2] | Mattei AL, Bailly N, Meissner A. DNA methylation: a historical perspective[J]. Trends Genet, 2022, 38(7): 676-707. | | [3] | Bewick AJ, Vogel KJ, Moore AJ, et al. Evolution of DNA methylation across insects[J]. Mol Biol Evol, 2017, 34(3): 654-665. | | [4] | Moore LD, Le T, Fan GP. DNA methylation and its basic function[J]. Neuropsychopharmacology, 2013, 38(1): 23-38. | | [5] | Dong YW, Hou JH, Zhu BC, et al. Concepts related to epigenetics and their advances[J]. J Biol, 2005, 22(1): 1-3. (in Chinese) | | | (董玉玮, 侯进慧, 朱必才, 等. 表观遗传学的相关概念和研究进展[J]. 生物学杂志, 2005, 22(1): 1-3.) | | [6] | Feng J, Chang H, Li E, et al. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system[J]. J Neurosci Res, 2005, 79(6): 734-746. | | [7] | Zhang GM, Hussain M, O’Neill SL, et al. Wolbachia uses a host microRNA to regulate transcripts of a methyltransferase, contributing to dengue virus inhibition in Aedes aegypti[J]. Proc Natl Acad Sci USA, 2013, 110(25): 10276-10281. | | [8] | Condé R, Hernandez-Torres E, Claudio-Piedras F, et al. Heat shock causes lower Plasmodium infection rates in Anopheles albimanus[J]. Front Immunol, 2021, 12: 584660. | | [9] | Claudio-Piedras F, Recio-Tótoro B, Condé R, et al. DNA methylation in Anopheles albimanus modulates the midgut immune response against Plasmodium berghei[J]. Front Immunol, 2020, 10: 3025. | | [10] | Oppold A, Kre? A, Vanden Bussche J, et al. Epigenetic alterations and decreasing insecticide sensitivity of the Asian tiger mosquito Aedes albopictus[J]. Ecotoxicol Environ Saf, 2015, 122: 45-53. | | [11] | Fonseca DM, Keyghobadi N, Malcolm CA, et al. Emerging vectors in the Culex pipiens complex[J]. Science, 2004, 303(5663): 1535-1538. | | [12] | Fonseca DM, Smith JL, Kim HC, et al. Population genetics of the mosquito Culex pipiens pallens reveals sex-linked asymmetric introgression by Culex quinquefasciatus[J]. Infect Genet Evol, 2009, 9(6): 1197-1203. | | [13] | Farajollahi A, Fonseca DM, Kramer LD, et al. “Bird biting” mosquitoes and human disease: a review of the role of Culex pipiens complex mosquitoes in epidemiology[J]. Infect Genet Evol, 2011, 11(7): 1577-1585. | | [14] | Eldridge BF. Diapause and related phenomena in Culex mosquitoes: their relation to arbovirus disease ecology[M]. New York, NY: Springer New York, 1987: 1-28. | | [15] | Wilton DP, Smith GC. Ovarian diapause in three geographic strains of Culex pipiens (Diptera ∶ Culicidae)[J]. J Med Entomol, 1985, 22(5): 524-528. | | [16] | Gomes B, Parreira R, Sousa CA, et al. The Culex pipiens complex in continental Portugal: distribution and genetic structure[J]. J Am Mosq Control Assoc, 2012, 28(4 Suppl): 75-80. | | [17] | Bittar E, Bittar N. Molecular and cellular genetics[J]. Elsevier, 1996: 33-66. | | [18] | Lyko F. DNA methylation learns to fly[J]. Trends Genet, 2001, 17(4): 169-172. | | [19] | Field LM, Lyko F, Mandrioli M, et al. DNA methylation in insects[J]. Insect Mol Biol, 2004, 13(2): 109-115. | | [20] | Shi RL, Jiang LL. Recent advances in peroxisomal fatty acid β-oxidation[J]. Chin J Biochem Mol Biol, 2009, 25(1): 12-16. (in Chinese) | | | (石如玲, 姜玲玲. 过氧化物酶体脂肪酸β氧化[J]. 中国生物化学与分子生物学报, 2009, 25(1): 12-16.) | | [21] | Campbell JA, Davies GJ, et al. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities[J]. Biochem J, 1998, 329 (Pt 3)(Pt 3):719. | | [22] | Lim EK, Bowles DJ. A class of plant glycosyltransferases involved in cellular homeostasis[J]. EMBO J, 2004, 23(15): 2915-2922. | | [23] | Yan DM, Shi GH, Li HJ, et al. Overwintering surveillance of Culex pipiens pallens in Shandong Province[J]. Chin J Schisto Control, 2018, 30(1): 65-67, 71. (in Chinese) | | | (严冬梅, 石桂红, 李怀菊, 等. 山东省淡色库蚊越冬情况调查[J]. 中国血吸虫病防治杂志, 2018, 30(1): 65-67, 71.) | | [24] | Robich RM, Denlinger DL. Diapause in the mosquito Culex pipiens evokes a metabolic switch from blood feeding to sugar gluttony[J]. Proc Natl Acad Sci U S A, 2005, 102(44): 15912-15917. | | [25] | Millar MJ, Fischer MI, Elcoate PV, et al. The effects of dietary zinc deficiency on the reproductive system of male rats[J]. Can J Biochem Physiol, 1958, 36(6): 557-569. | | [26] | Lang CA. The accumulation of zinc by the mosquito[J]. 1963, 46(3): 617-627. | | [27] | Zhao X, Smartt CT, Hillyer JF, et al. A novel member of the RING-finger gene family associated with reproductive tissues of the mosquito, Aaedes aegypti[J]. Insect Mol Biol, 2000, 9(3): 301-308. | | [28] | Su TY, Lu YR. Autogeny of mosquitoes[J]. Entomol Knowl, 1988, 25(4): 246-247. (in Chinese) | | | (苏天运, 卢艳如. 蚊虫的自育性[J]. 昆虫知识, 1988, 25(4): 246-247.) | | [29] | Tang CY, Zhao LH, Li ZY. Functions of N6-methyladenosine modification in viral infection[J]. Prog Microbiol Immunol, 2024, 52(2): 72-78. (in Chinese) | | | (唐成嵛, 赵兰华, 李忠玉. N-6-腺苷酸甲基化修饰与病毒感染的研究进展[J]. 微生物学免疫学进展, 2024, 52(2): 72-78.) | | [30] | Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq[J]. Nature, 2012, 485(7397): 201-206. | | [31] | Zamocky M, Furtmüller PG, Obinger C. Evolution of catalases from bacteria to humans[J]. Antioxid Redox Signal, 2008, 10(9): 1527-1548. | | [32] | Zhang KS, Tian HL. Research and function of catalase in organism[J]. Food Sci Technol, 2007, 32(1): 8-11. (in Chinese) | | | (张坤生, 田荟琳. 过氧化氢酶的功能及研究[J]. 食品科技, 2007, 32(1): 8-11.) | | [33] | Lu BL, Tan JX, Li LZ, et al. Observation on indoor invasion activities of common mosquito species at night in Mubian, Guangxi[J]. Acta Entomol Sin, 1961, 4(S1): 401-410. (in Chinese) | | | (陆宝麟, 谭璟宪, 李丽璋, 等. 广西睦边常见蚊种夜晚侵入室内活动的观察[J]. 昆虫学报, 1961, 4(S1): 401-410.) | | [34] | Lehman CW, Lee JDR, Komives CF. Ubiquitously expressed GPCR membrane-trafficking orthologs[J]. Genomics, 2005, 85(3): 386-391. |
|

 ), 黄新安1, 徐寒黎3, 李春晓1,2, 刘康康2, 邢丹2, 赵腾2,*(
), 黄新安1, 徐寒黎3, 李春晓1,2, 刘康康2, 邢丹2, 赵腾2,*(