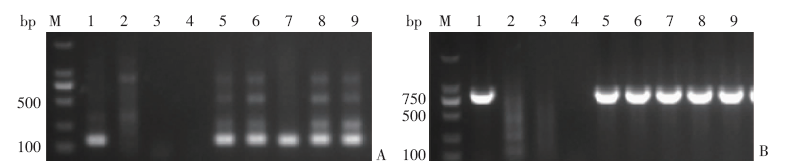
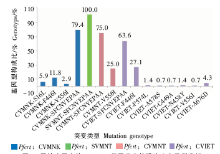
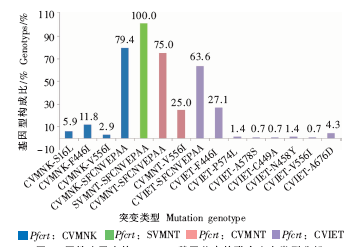
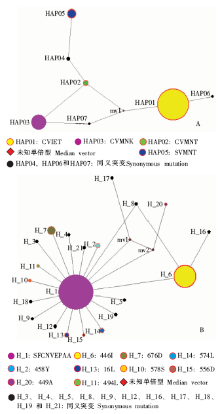
| [1] |
车立刚, 黄开国, 杨恒林. 抗氯喹恶性疟原虫株对咯萘啶敏感性的测定[J]. 中国寄生虫学与寄生虫病杂志, 1984, 2(4): 280.
|
| [2] |
杨恒林, 杨品芳, 董莹, 等. 云南省恶性疟原虫对氯喹抗性的纵向监测[J]. 中国寄生虫学与寄生虫病杂志, 1994, 2(1): 31-33.
|
| [3] |
杨恒林, 李春富, 杨亚明, 等. 云南东南部地区恶性疟原虫对氯喹敏感性纵向观察[J]. 中国病原生物学杂志, 2008, 3(12): 917-919.
|
| [4] |
车立刚, 陈文才, 杨恒林, 等. 云南省恶性疟原虫对氯喹抗性的消长[J]. 中华流行病学杂志, 1986 (7): 88-91.
|
| [5] |
车立刚. 新抗疟药“224”治疗恶性疟疾65例的效果观察[J]. 云南医药, 1981(6): 40-43.
|
| [6] |
王同寅, 唐宝璋, 徐汝昌, 等. 青蒿素衍生物“224”与“242”治疗恶性疟的临床研究[J]. 昆明医科大学学报, 1981(2): 56-58.
|
| [7] |
王同寅. 青蒿素制剂治疗疟疾的临床研究[J]. 中国医刊, 1981(7): 19-20.
|
| [8] |
郭兴伯. 青蒿素治疗脑型疟疾36例小结[J]. 中医杂志, 1979(1): 56-58.
|
| [9] |
郭兴伯, 符林春, 简华香, 等. 青蒿素栓治疗凶险型疟疾32例临床报告[J]. 广州中医药大学学报, 1986(4): 15-17.
|
| [10] |
广东中医学院五二三海南工作组. 青蒿素治疗抗氯喹疟疾36例效果观察[J]. 广东卫生防疫, 1979, 1: 50-56.
|
| [11] |
刘德全, 冯晓平, 杨恒林, 等. 我国恶性疟原虫对氯喹抗性的消长[J]. 中国寄生虫学与寄生虫病杂志, 2005, 23(1): 27-31.
|
| [12] |
刘德全, 刘瑞君, 张春勇, 等. 我国恶性疟原虫对抗疟药敏感性的现状[J]. 中国寄生虫学与寄生虫病杂志, 1996, 14(1): 39-43.
|
| [13] |
杨恒林, 李兴亮, 高白荷, 等. 中缅边境西段恶性疟原虫对7种抗疟药敏感性的监测[J]. 中国病原生物学杂志, 2009, 4(11): 831-832, 843.
|
| [14] |
朱垚吉, 陈梦妮, 徐艳春, 等. 云南省恶性疟原虫Pfcrt基因exon2区72~76编码序列多态性的分析[J]. 中国寄生虫学与寄生虫病杂志, 2016, 34(3): 227-232, 234.
|
| [15] |
孙艾明, 董莹, 陈梦妮, 等. 云南省恶性疟原虫青蒿素耐药性相关基因K13 kelch结构域序列多态性的分析[J]. 中国寄生虫学与寄生虫病杂志, 2016, 34(4): 339-345.
|
| [16] |
Wernsdorfer WH, Landgraf B, Wiedermann G, et al. Inverse correlation of sensitivity in vitro of Plasmodium falciparum to chloroquine and mefloquine in Ghana[J]. Trans R Soc Trop Med Hyg, 1994, 88(4): 443-444.
|
| [17] |
Wernsdorfer WH, Landgraf B, Wiedermann G, et al. Chloroquine resistance of Plasmodium falciparum: a biological advantage[J]. Trans R Soc Trop Med Hyg, 1995, 89(1): 90-91.
|
| [18] |
Wongsrichanalai C, Pickard AL, Wernsdorfer WH, et al. Epidemiology of drug-resistant malaria[J]. Lancet Infect Dis, 2002, 2(4): 209-218.
|
| [19] |
Wootton JC, Feng X, Ferdig MT, et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum[J]. Nature, 2002, 418(6895): 320-323.
|
| [20] |
Kateera F, Nsobya SL, Tukwasibwe S, et al. Molecular surveillance of Plasmodium falciparum drug resistance markers reveals partial recovery of chloroquine susceptibility but sustained sulfadoxine-pyrimethamine resistance at two sites of different malaria transmission intensities in Rwanda[J]. Acta Tropica, 2016, 164: 329-336.
|
| [21] |
Menegon M, Talha AA, Severini C, et al. Frequency distribution of antimalarial drug resistance alleles among Plasmodium falciparum isolates from Gezira State, central Sudan, and Gedarif State, eastern Sudan[J]. Am J Trop Med Hyg, 2010, 83(2): 250-257.
|
| [22] |
Boussaroque A, Fall B, Madamet M, et al. Prevalence of anti-malarial resistance genes in Dakar, Senegal from 2013 to 2014[J]. Malar J, 2016, 15(1): 347.
|
| [23] |
Nyunt MH, Hlaing T, Oo HW, et al. Molecular assessment of artemisinin-resistance markers, polymorphisms in the K13 propeller and a multidrug-resistance gene, in eastern and western border areas of Myanmar[J]. Clin Infect Dis, 2015, 60(8): 1208-1215.
|
| [24] |
Talundzic E, Okoth SA, Congpuong K, et al. Selection and spread of artemisinin-resistant alleles in Thailand prior to the global artemisinin resistance containment campaign[J]. PLoS Pathog, 2015, 11(4): e1004789.
|
| [25] |
Mita T, Venkatesan M, Ohashi J, et al. Limited geographical origin and global spread of sulfadoxine-resistant dhps alleles in Plasmodium falciparum populations[J]. J Infect Dis, 2011, 204(12): 1980-1988.
|
| [26] |
Alam MT, de Souza DK, Vinayak S, et al. Selective sweeps and genetic lineages of Plasmodium falciparum drug-resistant alleles in Ghana[J]. J Infect Dis, 2011, 203(2): 220-227.
|
| [27] |
中华人民共和国卫生部. 中华人民共和国卫生行业标准: 疟疾的诊断(WS259-2015)[S]. 北京: 人民卫生出版社, 2006: 2-3.
|
| [28] |
董莹, 毛祥华, 陈梦妮, 等. 2012-2014年云南省疟疾实验室诊断质量分析[J]. 中国寄生虫学与寄生虫病杂志, 2015, 33(3): 191-195.
|
| [29] |
Lai MT, Lu M, Felock PJ, et al. Distinct mutation pathways of non-subtype B HIV-1 during in vitro resistance selection with nonnucleoside reverse transcriptase inhibitors[J]. Antimicrob Agents Chemother, 2010, 54(11): 4812-4824.
|
| [30] |
Shen Y, Zhu YM, Fan X, et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia[J]. Blood, 2011, 118(20): 5593-5603.
|
| [31] |
沈杨, 陈赛娟. 急性白血病基因突变与多步骤发病机制和临床相关性[J]. 中国科学: 生命科学, 2013, 43(1): 39-45.
|
| [32] |
Chen D, Li PW, Goldstein BA, et al. Germline signaling mediates the synergistically prolonged longevity produced by double mutations in daf-2 and rsks-1 in C. elegans[J]. Cell Rep, 2013, 5(6): 1600-1610.
|
| [33] |
武菲, 胡文革, 王翠华, 等. 河鲈养殖与野生群体遗传多样性比较分析[J]. 水生生物学报, 2016, 40(1): 181-188.
|
| [34] |
周晓农, 张少森, 徐俊芳, 等. 我国消除疟疾风险评估分析[J]. 中国寄生虫学与寄生虫病杂志, 2014, 32(6): 414-418.
|

 ), 孙艾明1,2, 邓艳1, 陈梦妮1, 徐艳春1, 毛祥华1
), 孙艾明1,2, 邓艳1, 陈梦妮1, 徐艳春1, 毛祥华1