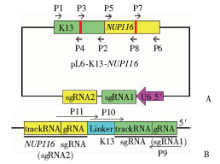
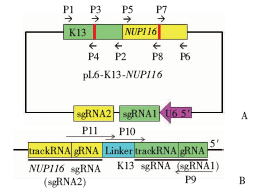
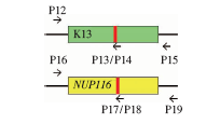
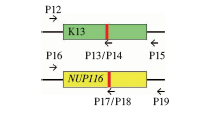
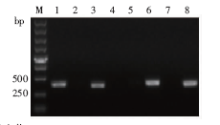
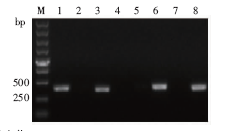
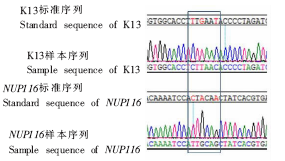
| [1] |
李奔福, 蔺应学, 郭祥瑞, 等. 中缅边境疟疾流行情况调查[J]. 中国寄生虫学与寄生虫病杂志, 2015, 33(4): 261-263.
|
| [2] |
尹授钦, 夏尚, 周兴武, 等. 云南省边境地区疟疾传播风险评估[J]. 中国寄生虫学与寄生虫病杂志, 2016, 34(3): 255-260.
|
| [3] |
张丽, 丰俊, 张少森, 等. 2015年全国疟疾疫情分析[J]. 中国寄生虫学与寄生虫病杂志, 2016, 34(6): 477-481.
|
| [4] |
雷蕾, 买买提江·吾买尔, 李志宏, 等. 输入性疟疾引起继发传播的风险评估研究进展[J]. 中国寄生虫学与寄生虫病杂志, 2016, 34(5): 468-472.
|
| [5] |
Noedl H, Se Y, Schaecher K, et al. Evidence of artemisinin-resistant malaria in western Cambodia[J]. N Engl J Med, 2008, 359(24): 2619-2620.
|
| [6] |
Fairhurst RM, Dondorp AM.Artemisinin-resistant Plasmodium falciparum malaria[J]. Microbiol Spectr, 2016, 4(3): 1-16.
|
| [7] |
张逸龙, 潘卫庆. 恶性疟原虫对青蒿素产生抗性的研究进展[J]. 中国寄生虫学与寄生虫病杂志, 2015, 33(6): 418-424.
|
| [8] |
Bhaya D, Davison M, Barrangou R.CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation[J]. Annu Rev Genet, 2011, 45: 273-297.
|
| [9] |
Pennisi E.The CRISPR craze[J]. Science, 2013, 341(6148): 833-836.
|
| [10] |
Barrangou R.RNA events. Cas9 targeting and the CRISPR revolution[J]. Science, 2014, 344(6185): 707-708.
|
| [11] |
Bawankar P, Shaw PJ, Sardana R, et al. 5′and 3′end modifications of spliceosomal RNAs in Plasmodium falciparum[J]. Mol Biol Rep, 2010, 37(4): 2125-2133.
|
| [12] |
Port F, Bullock SL.Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs[J]. Nat Methods, 2016, 13(10): 852-854.
|
| [13] |
Kennedy EM, Cullen BR. Bacterial CRISPR/Cas DNA endonuc-leases: a revolutionary technology that could dramatically impact viral research and treatment[J]. Virology, 2015, 479-480: 213-220.
|
| [14] |
Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J]. Science, 2012, 337(6096): 816-821.
|
| [15] |
Ghorbal M, Gorman M, Macpherson CR, et al. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system[J]. Nat Biotechnol, 2014, 32(8): 819-821.
|
| [16] |
Wagner JC, Platt RJ, Goldfless SJ, et al. Efficient CRISPR-Cas9-mediated genome editing in Plasmodium falciparum[J]. Nat Methods, 2014, 11(9): 915-918.
|
| [17] |
Li P, Estrada JL, Burlak C, et al. Efficient generation of genetically distinct pigs in a single pregnancy using multiplexed single-guide RNA and carbohydrate selection[J]. Xenotransplantation, 2015, 22(1): 20-31.
|
| [18] |
Nissim L, Perli SD, Fridkin A, et al. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells[J]. Mol Cell, 2014, 54(4): 698-710.
|
| [19] |
Wang H, Yang H, Shivalila CS, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering[J]. Cell, 2013, 153(4): 910-918.
|

 )
)